Spectrum preparation
After the initial (predicted) values (J-couplings, chemical shifts, etc.) for the spin system are set up, the spin system optimization can begin. Some spectrum preparation should be done before starting the iterative analysis. The following contains a basic protocol with essential steps for spectrum preparation.
-
Simulate the spectrum by following QMSA → Simulate (or by pressing Ctrl + R).
-
Do baseline correction if clearly positive or negative baseline.
-
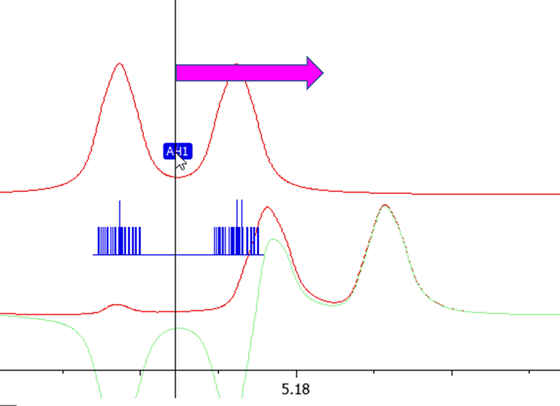
Adjust the positions of the chemical shifts by dragging the shifts with left mouse and by matching the chemical shifts and resonance patterns of each signal to the observed spectrum.
-
Simulate spectrum. It is sensible to save the parameter file now. Select File → Save “filename” As…
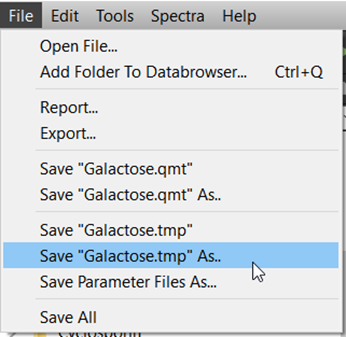
-
Select the file extension for the parameter file.
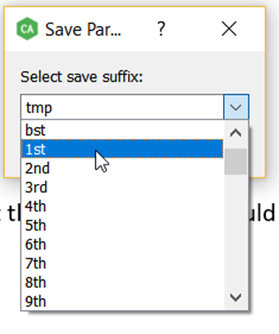
-
Check that the given parameters and the predicted ones are not very different and that their ranges are realistic. At the beginning of the analysis ranges should be large enough (1 ppm for shifts, 4-10 Hz for couplings) or they could be completely ignored (see ranges of spectral parameters).
-
Check that the populations, coupling constants and chemical shifts are optimizable (see fixing and unfixing parameters). It its recommended to keep response factors always fixed.
-
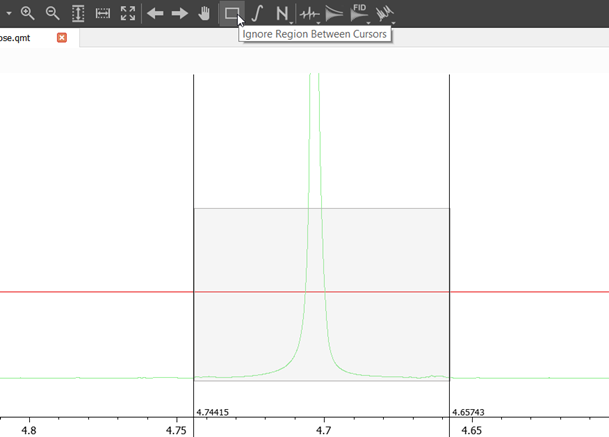
Remove/cover solvent and other extra signals via Ignore Region Between Cursors button, as shown in the image below.