Tutorials
Advanced tutorial 1
In this tutorial we analyze quantitatively a mixture of compounds with ChemAdder.
Contents
Initialization
Now we have very good quality spectrum and we don't need to correct the phase or baseline.
Let's start by initializing the parameters of used reference and solvent. Click on the 'Spin Systems' tab the 'Add' button and choose 'Add Reference...':

Add the desired reference, this time it's TSP with the satellite signals:
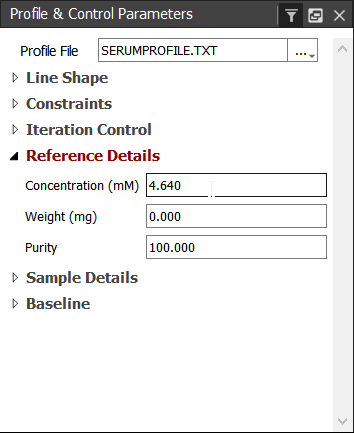
Remember to check also the concentration of the reference. The default concentration (4.64 mmol/l) is correct at this time:
Next we can add the used solvent. This is crucial if some of the signals are overlapping with the solvent signal. This is not the case, but let's practise it, though. Click on the 'Spin Systems' tab the 'Add' button and choose 'Add Solvents...':
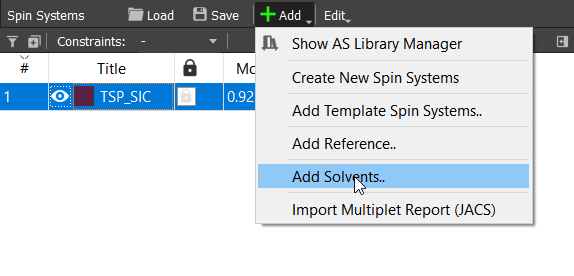
Choose 'D2O' from the list. This will add residual signal of HDO.
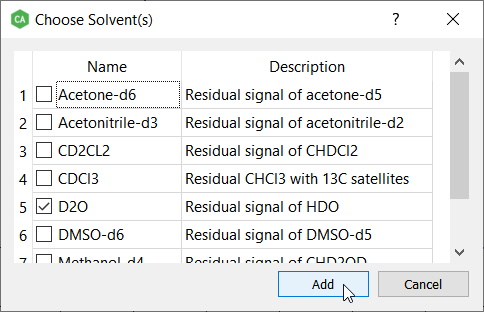
If the solvent signal isn't in the right position, correct the position. It's always good idea to iterate the spectrum after adding the solvent and reference spin systems.
Compounds
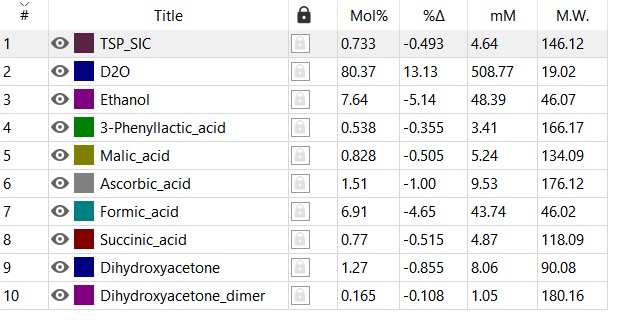
We have a mixture of formic acid, succinic acid (butanedioic acid), malic acid (2-hydroxybutanedioic acid), 1,3-dihydroxyacetone, 3-phenyllactic acid, ascorbic acid and ethanol.
Analyzation
Now we can start by integrating the signals to identify the spin systems.
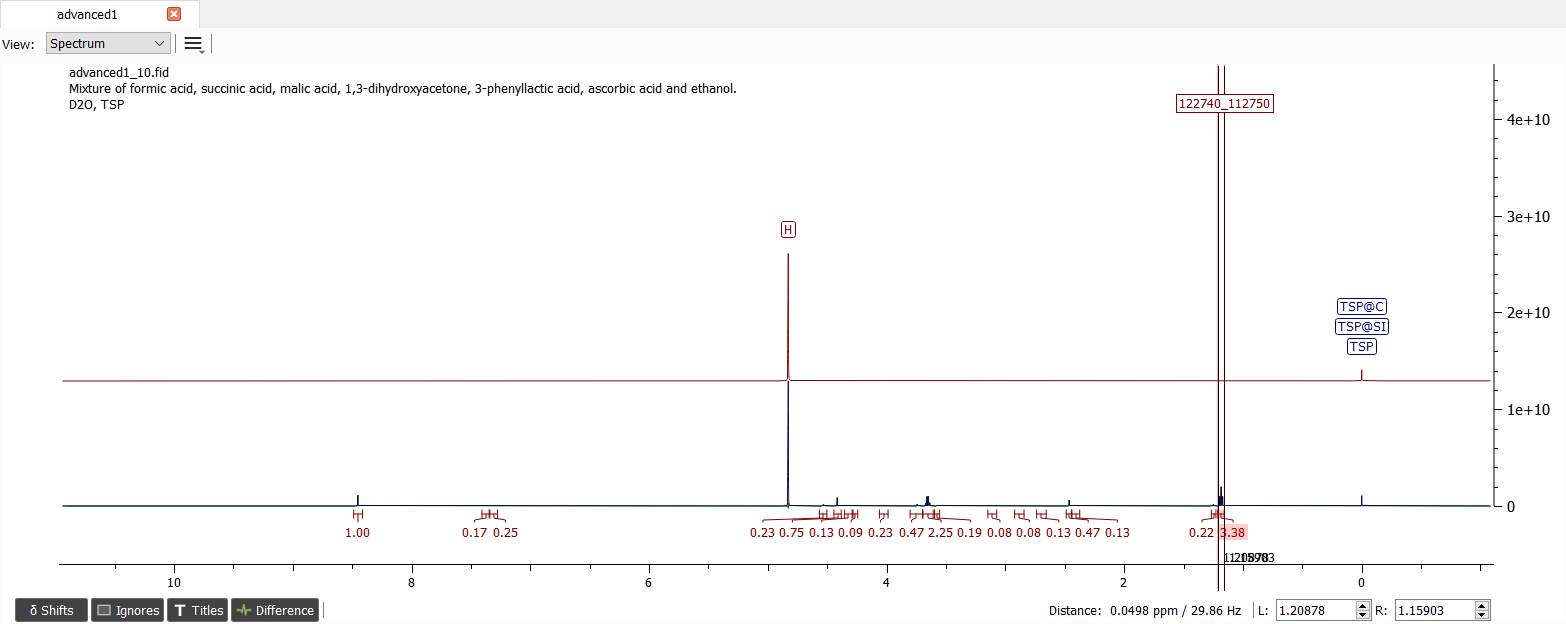
Let's normalize the largest integral (at ~1.18 ppm). Because the chemical shift is quite traditional for methyl group, let's normalize the signal to three. At ~3.65 ppm there is another signal which integral is close to an integer of two and the splitting pattern (quartet) is very familiar to us so these two signals correspond to ethyl group. The chemical shift value indicates also vicinity of hydroxyl group so our first compound is ethanol. Note that D2O prevents the hydroxyl signal to be seen in the spectrum. Let's create the first spin system for this molecule. Edit the spin particle types and guess the J-coupling. After iteration the spectrum should look like this:
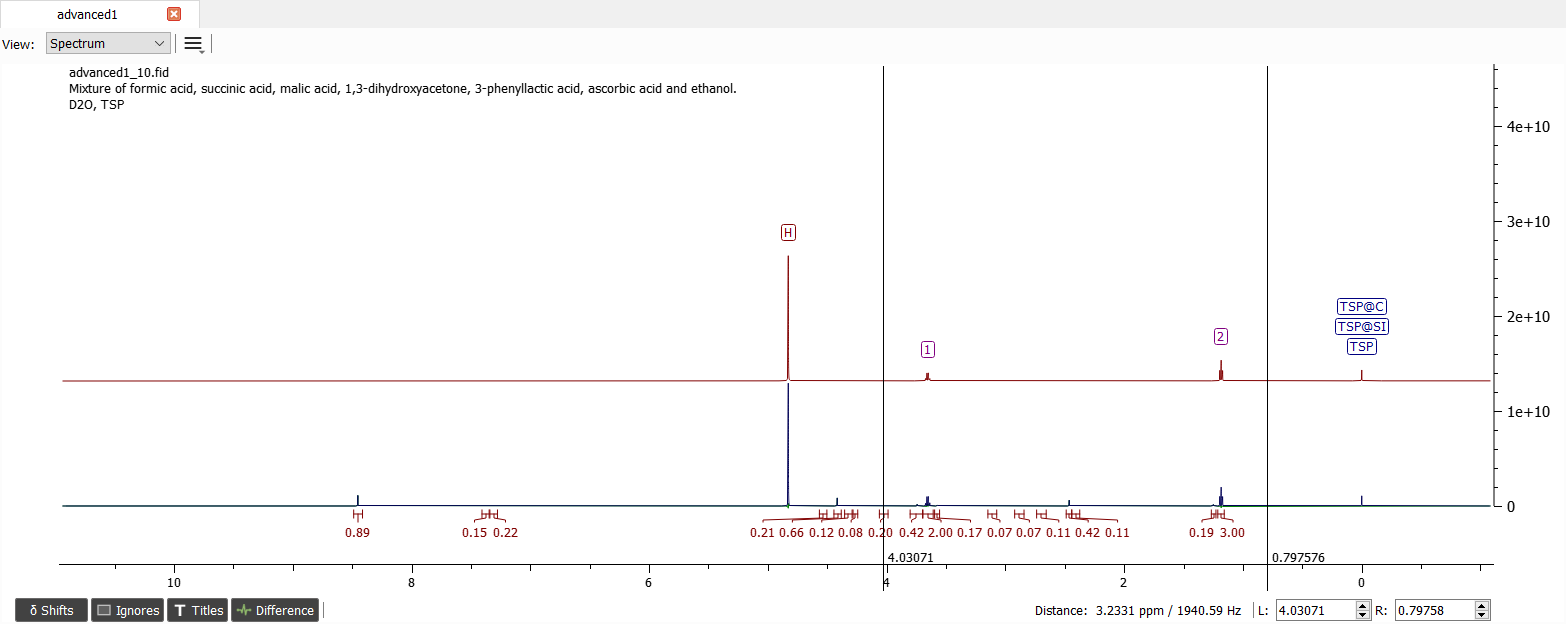
Another very diagnostic region is the aromatic region. Obviously these are the signals of 3-phenyllactic acid. Let's normalize the left integral to two in the aromatic region and the whole spectrum should look like this:
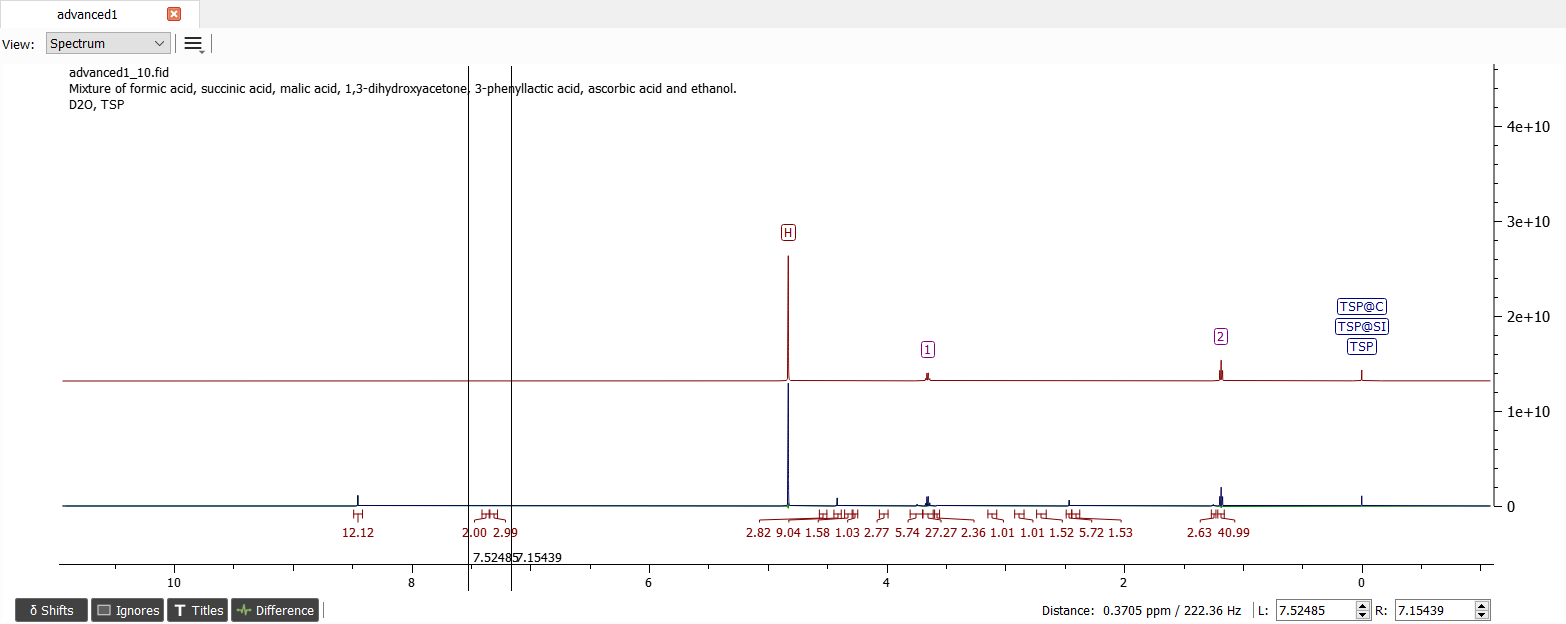
Note the three signals with the integral values of about one. Now we can create spin system with six chemical shifts. Let's start analyzing the aromatic region. After editing spin particle types (1: '1*2*1', 2: '1*1*1' and 3: '1*2*1') we can guess the J-couplings between these signals. We can notice from the splitting pattern that the shift '1' belongs to protons in meta position. Guesses for the couplings can be e.g.:
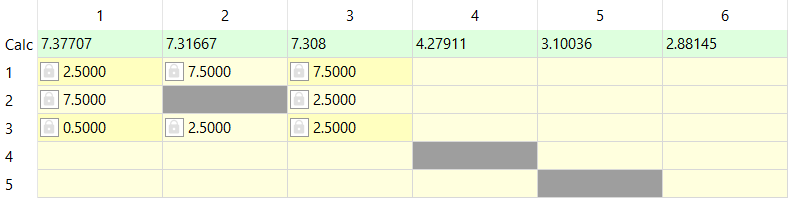
We can now simulate the spectrum and the spectrum should look like this:
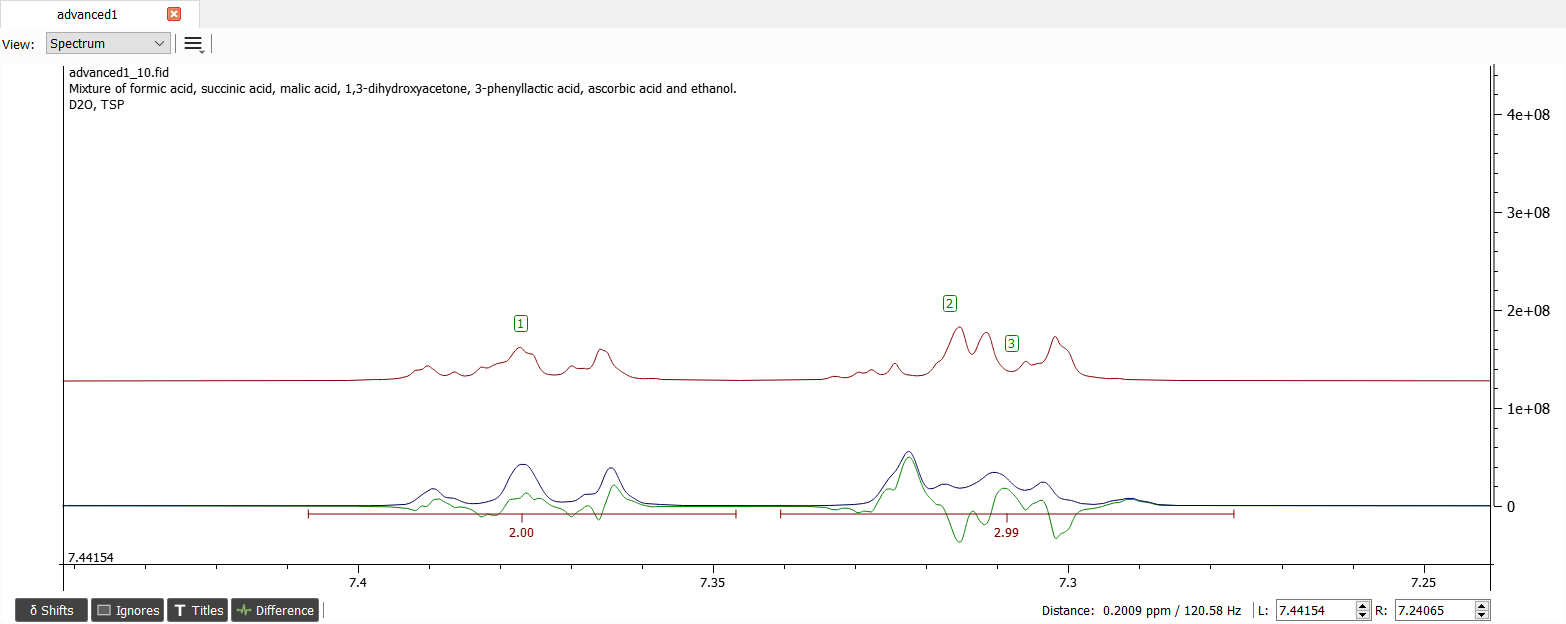
It seems that the shifts '2' and '3' are in wrong order. We can either manually drag the shifts to correct positions or just swap the shifts '2' and '3' by selecting the shifts and right-clicking and choosing 'Interchange/Swap Shifts'. After simulation the spectrum should look like this:
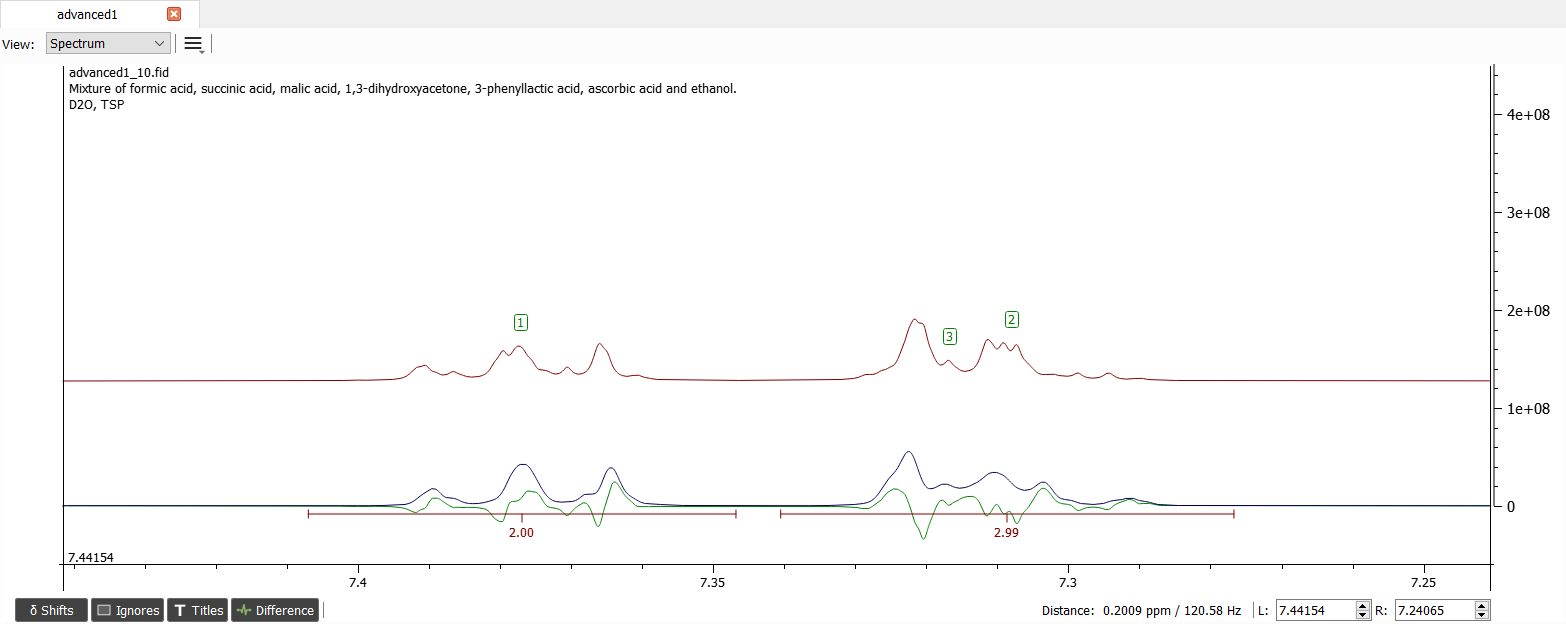
Now the signals seem to have quite correct parameters. Let's iterate until convergence and the spectrum should look like this:
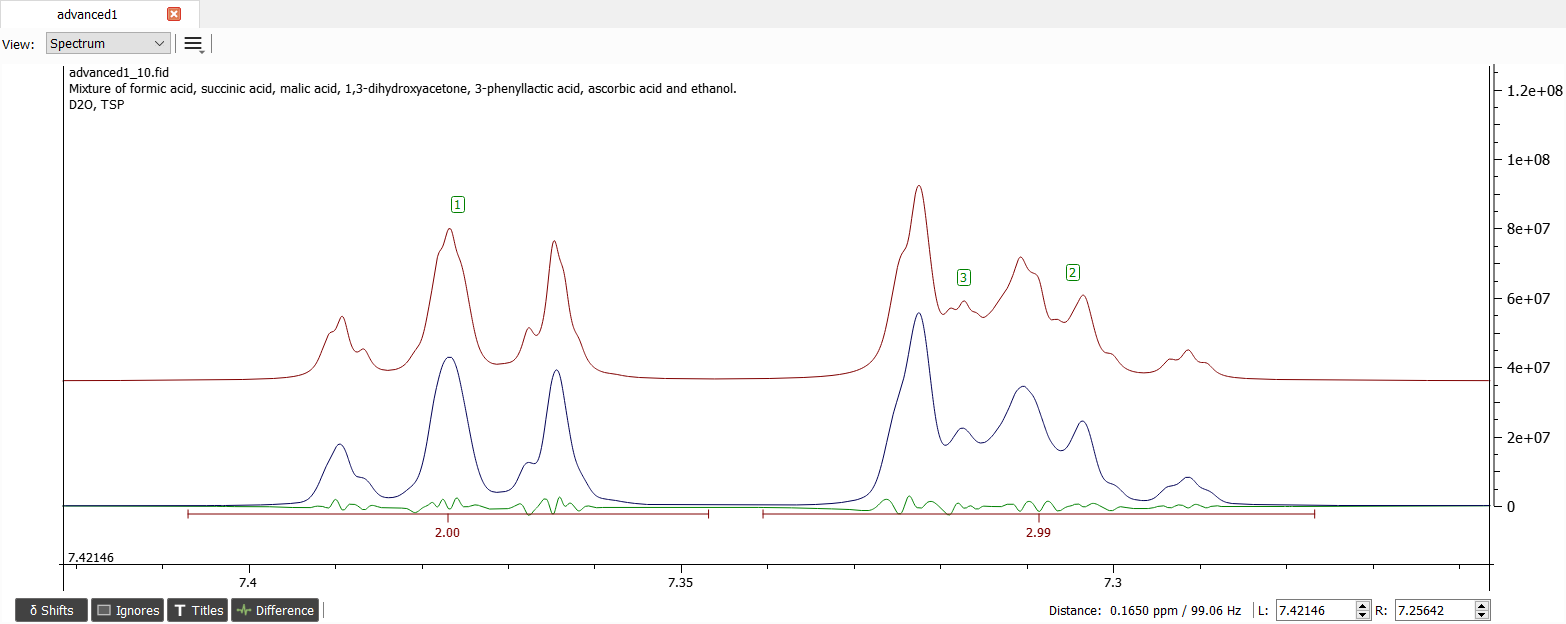
Now the aromatic region seems to be fine and we can continue with rest the of signals at 4.27, 3.11 and 2.88 ppm with labels '4', '5' and '6' respectively. The right ones seems to be coupled strongly with about 14 Hz. Note that this indicates they are geminal protons so the value should be -14 Hz. Also the signals seem to be coupled with each other. The coupling between '4' and '5' is about 4.3 Hz and the coupling between '4' and '6' is about 8 Hz. Note that we may need to lower the 'PCR Threshold' to value of 0.1 or lower. If the value is set exactly to 0, then another procedure is used.
After iteration the spectrum should look like this:
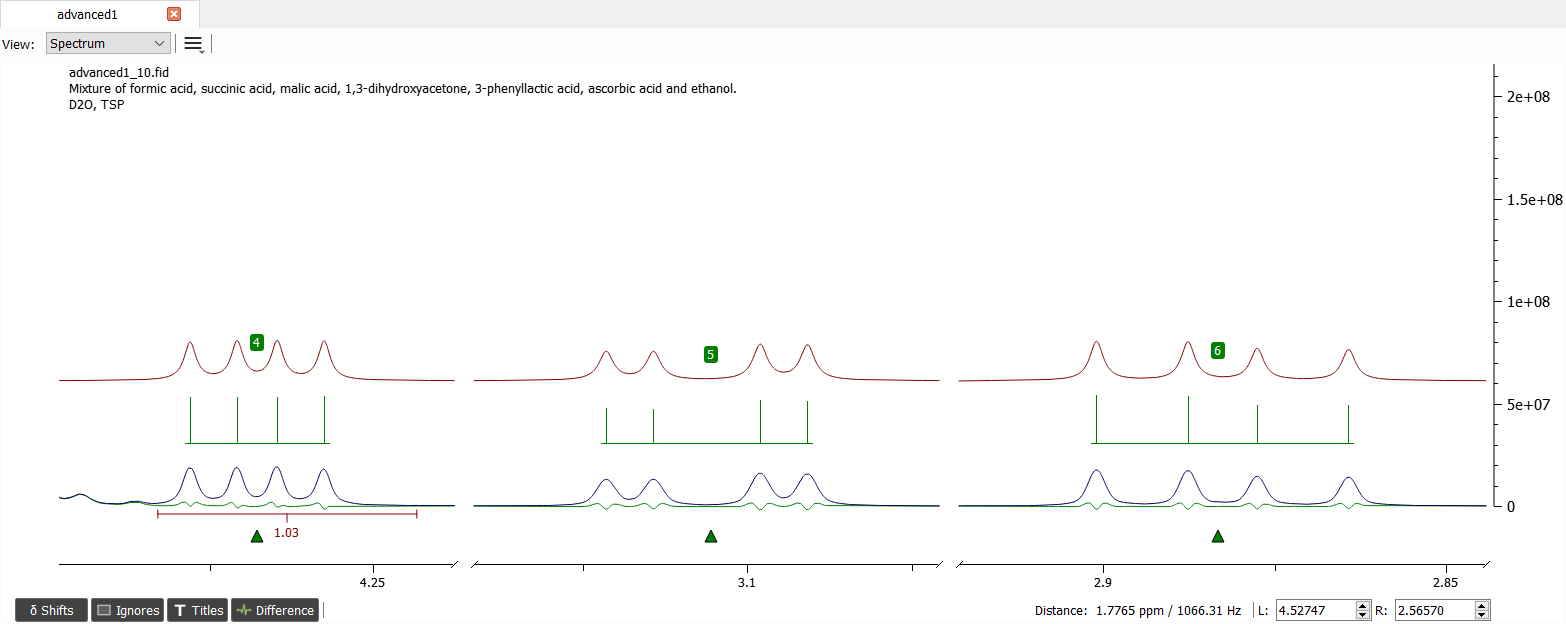
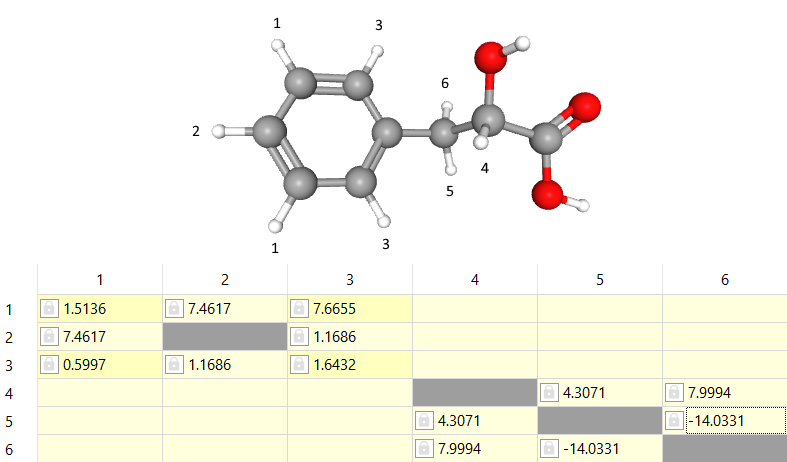
The assigned shifts and J-couplings are in the picture below:
There are quite similar chemical shifts close to the previous shifts '4' and '6'. After normalized integrals the spectum looks like this:
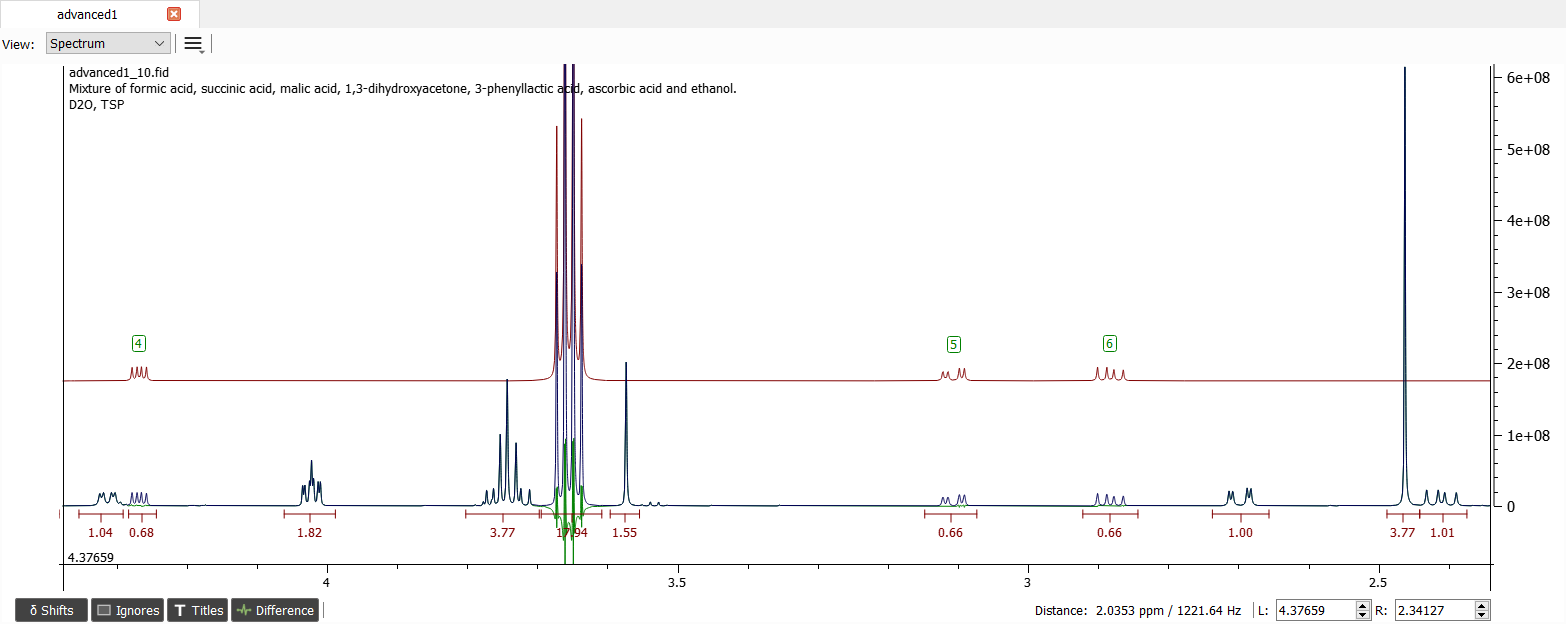
These signals belong to malic acid. After creating spin system and iterating, the parameters for these signals are:
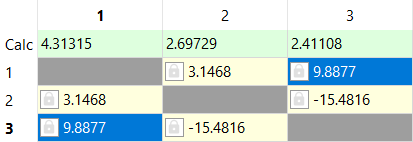
The assigned shifts are:
Let's create spin system for ascorbic acid next. There are four shifts which are located approximately at 4.535, 4.02, 3.755 and 3.73 ppm.
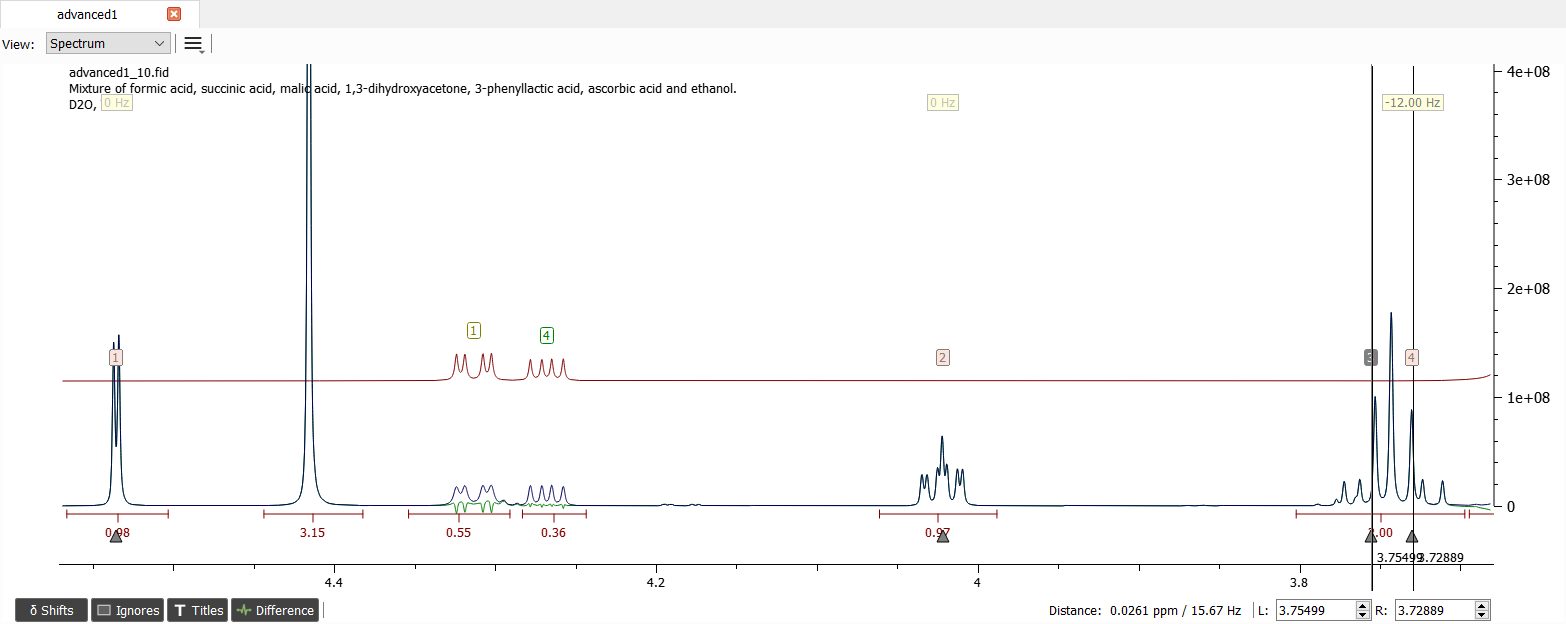
After adding guesses for the J-couplings and iteration, the spectrum looks like this:
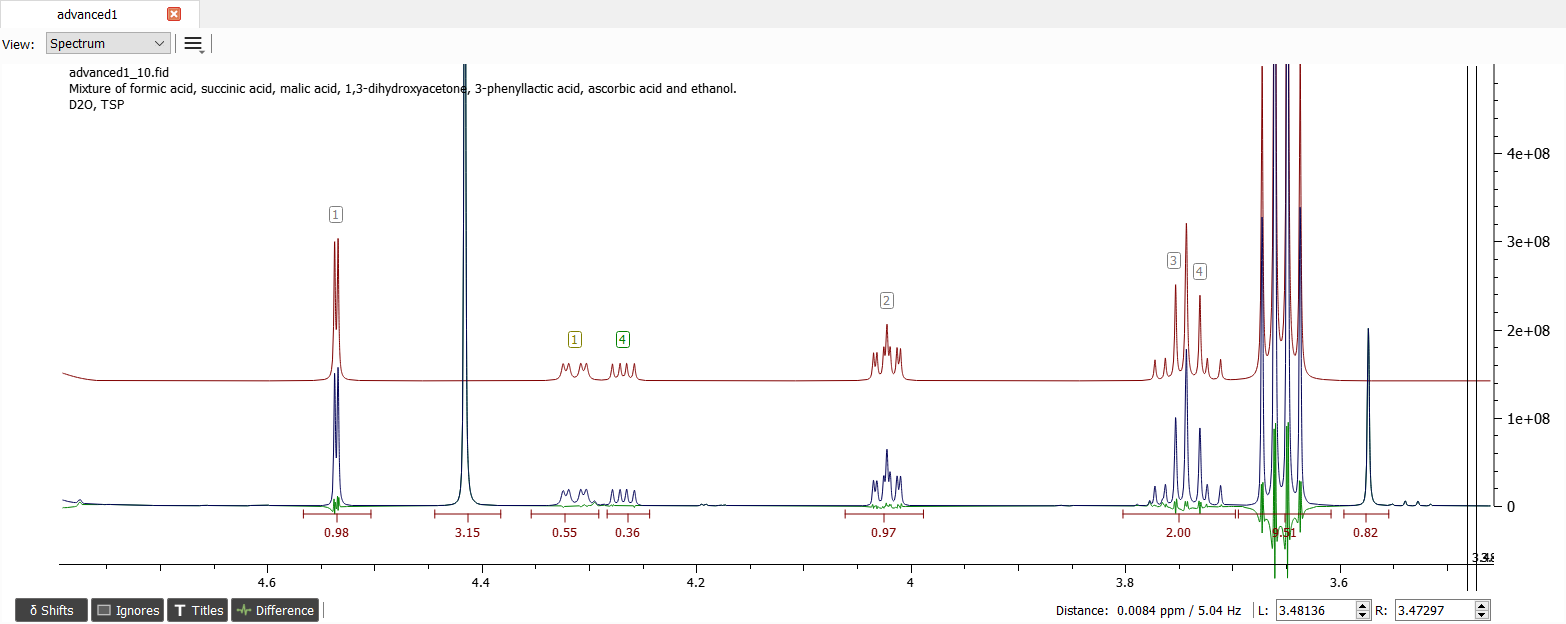
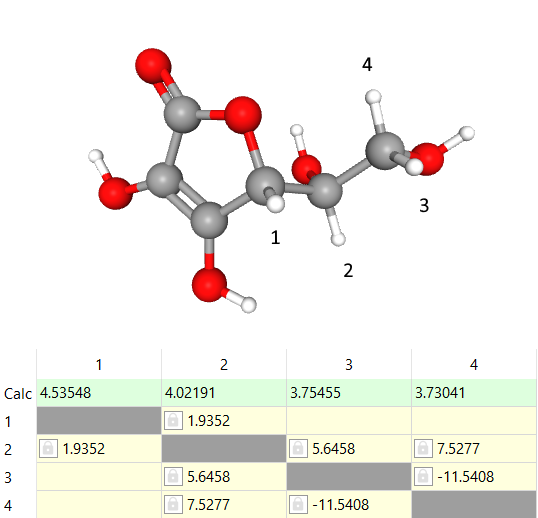
The assigned shifts and J-couplings of ascorbic acid are in the picture below:
Now we have five signals and only three compounds left which have one signal on the basis of the structures. The easiest signal is at 8.45 ppm which belongs to formic acid.
Next one is succinic acid which has signal at 2.46 ppm. The last compound is 1,3-dihydroxyacetone which has signal at 4.415 ppm. Dihydroxyacetone also forms dimer which has signal at 3.573 ppm:
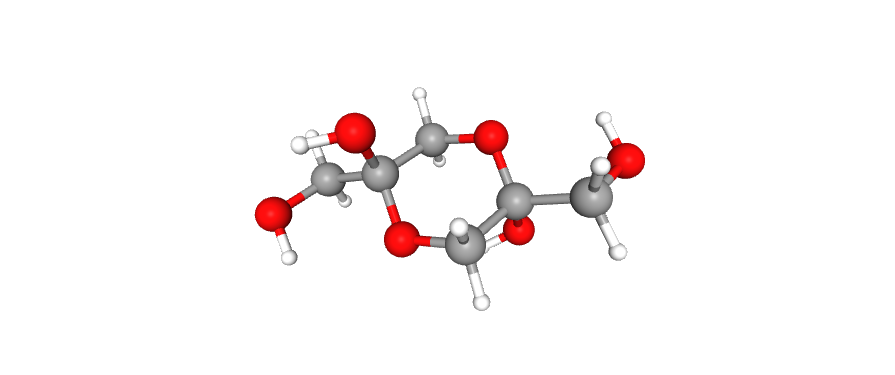
Dimerization process is so rapid that all of the protons of dimer are chemically equivalent.
The last signal 1.25 ppm remains unclear and can be ignored. The last phase is line shape optimization. After line shape optimization the RMS and R2 should be close to:

After converged RMS and R2 the concentrations of compounds are: