Tutorials
Advanced tutorial 2
In this tutorial we will use ChemAdder to determine concentrations of the two most abundant glucose isomers: β-glucopyranose and α-glucopyranose. There is also small amounts of β-glucofuranose, α-glucofuranose and open-chain form of glucose.
Contents
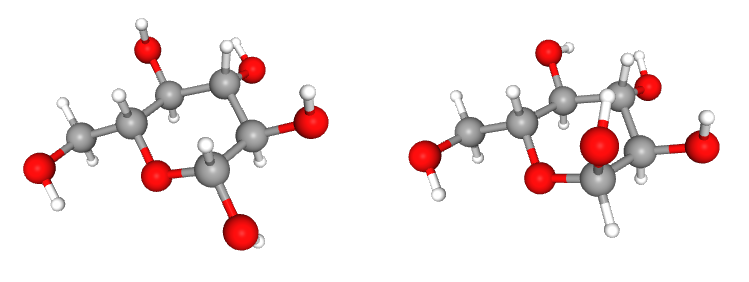
Molecule structures
The two molecule structures we are interested in (left: β-glucopyranose and right: α-glucopyranose):
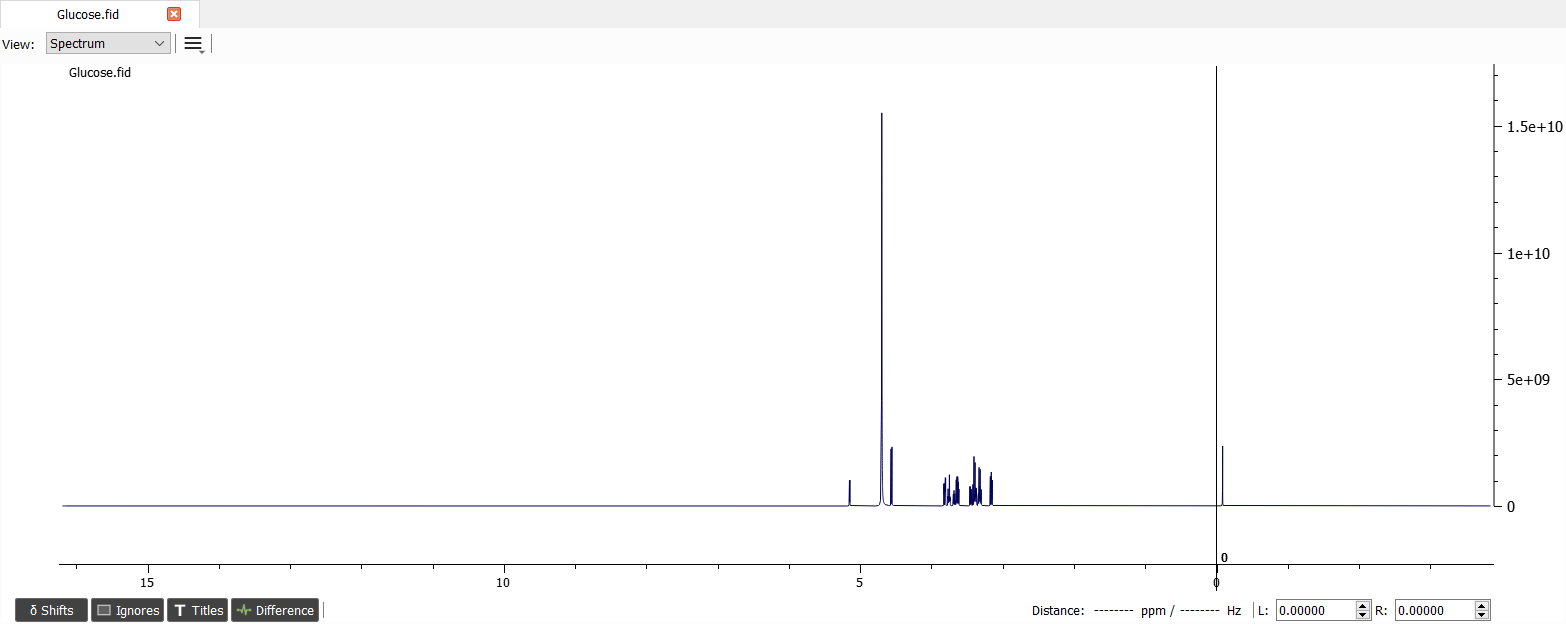
Carbohydrates have very specific proton: anomeric proton. The chemical shift of anomeric proton (next to the glycosidic bond) is located at 4.3 - 5.9 ppm. The anomeric proton signals can be seen next to the water signal:
Phase and baseline correction
Let's start with correcting phase of the spectrum.
The phase corrected spectrum will look like this:
The baseline correction can be made with 'Crowded Whittaker Smoothing' and filter of 230:
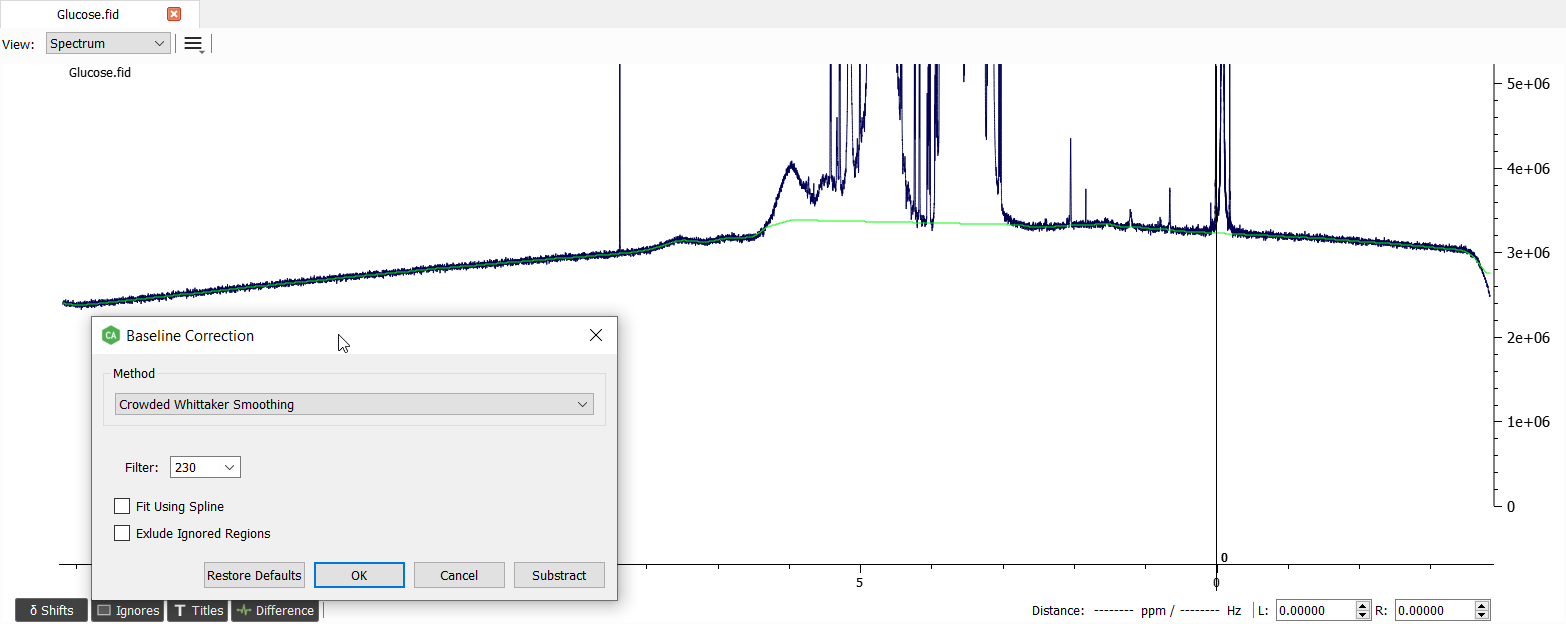
The reference signal (TSP) seems to be a bit off. Let's correct the position by setting the marginal position on the reference signal and choosing 'Reference Scale':
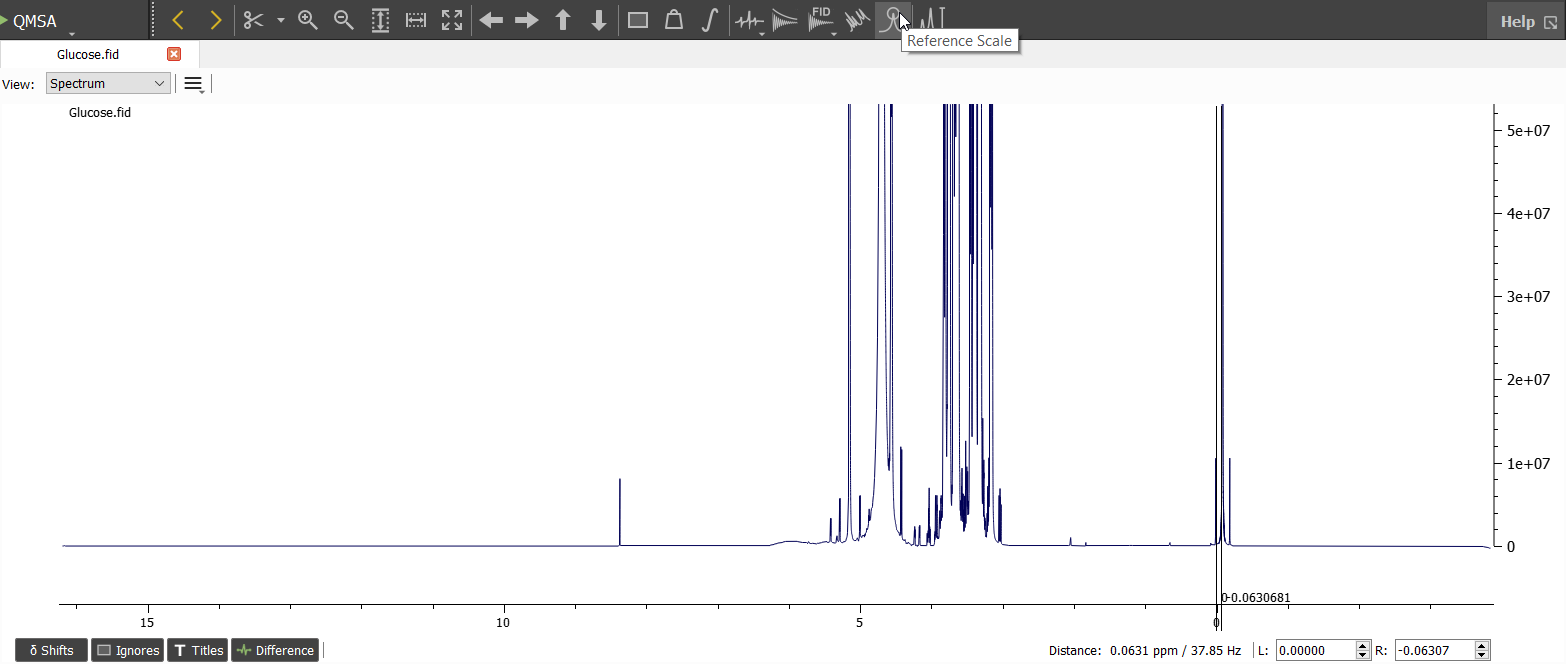
We can zoom out to see that the algorithm finds the highest point of the signal:
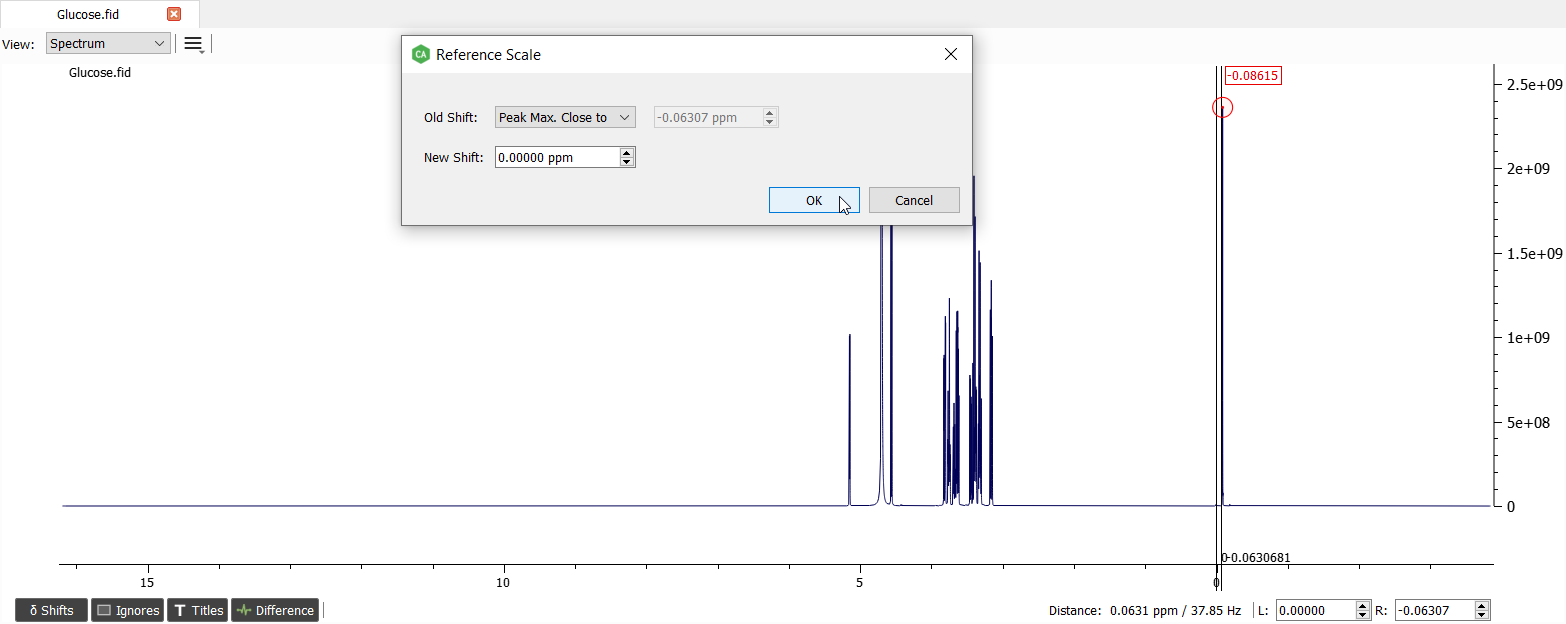
We want the reference signal to be at 0 ppm so we can just click Ok.
Initialization
Add TSP reference with concentration of 4.64 mmol/l and D2O solvent like in the previous tutorial. Move the solvent signal to correct position and iterate the spectrum:
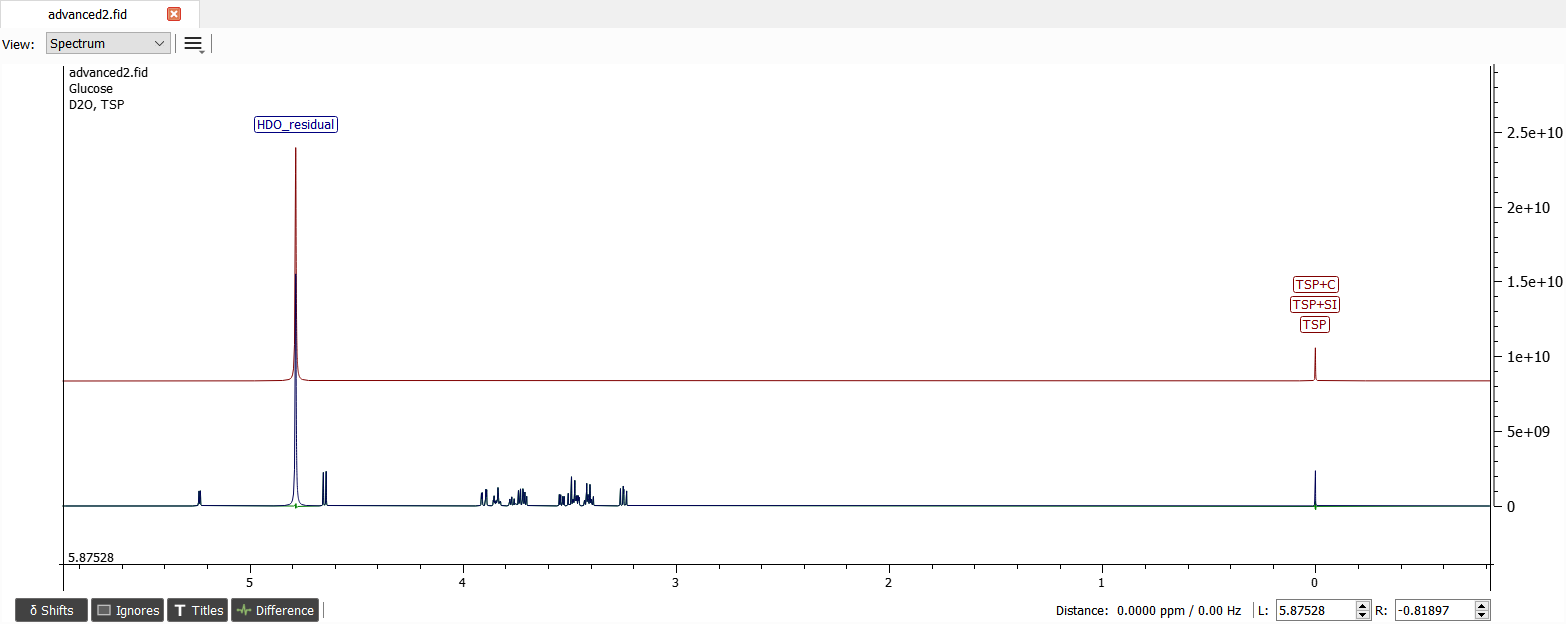
Analyzation
Now we know the molecules are β-glucopyranose and α-glucopyranose. Let's add these molecule structures (B-Glucose.sdf and A-Glucose.sdf) from 'Project Tree' by double-clicking or dragging to the 'Spectrum Window'. Note that the file filter needs to be either 'All Files (*)' or 'Chemical Structures (*.sdf *.mol)'. These molecule structures appears to the top of each other but they can moved by dragging. Now we can predict the Nmr parameters by right-clicking the structures and selecting 'NMR Prediction...'.
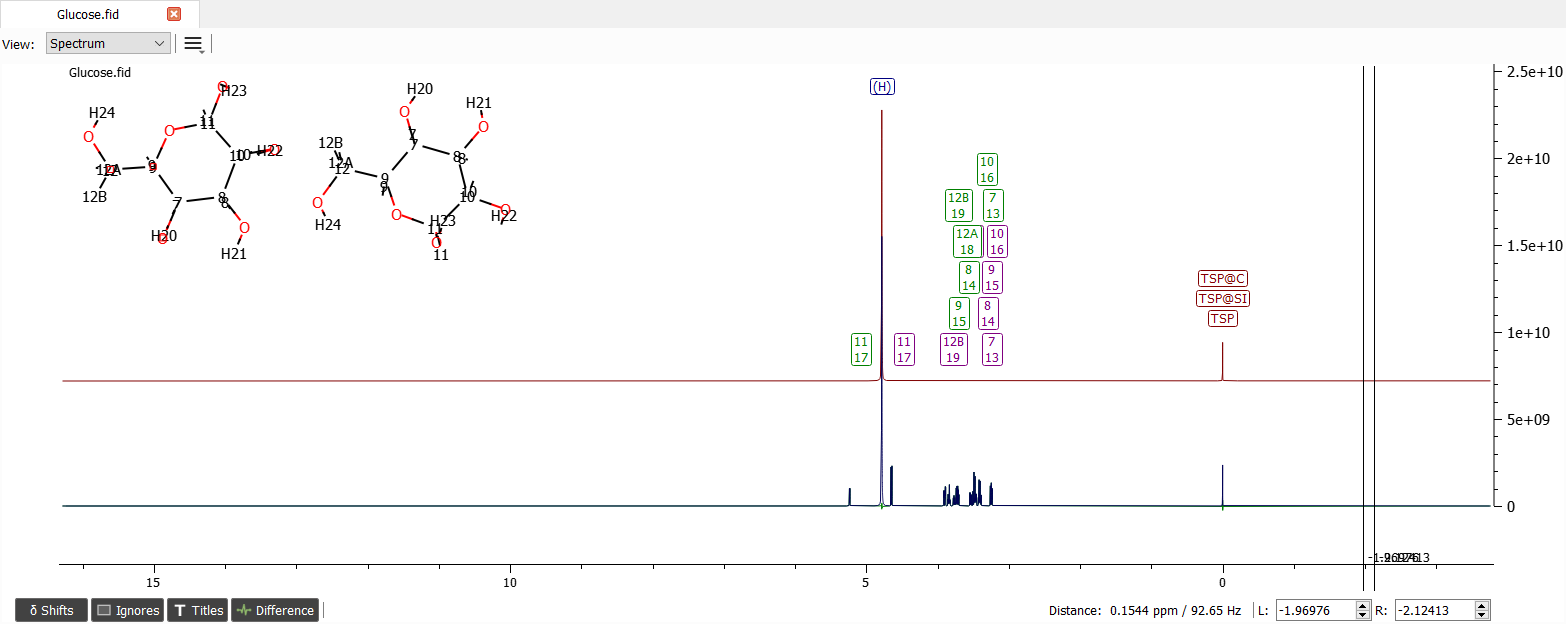
We may note that the predicted values aren't perfect but we may trust that the anomeric protons shifts are in right order. These can be moved to right positions. The spectrum can also be integrated:
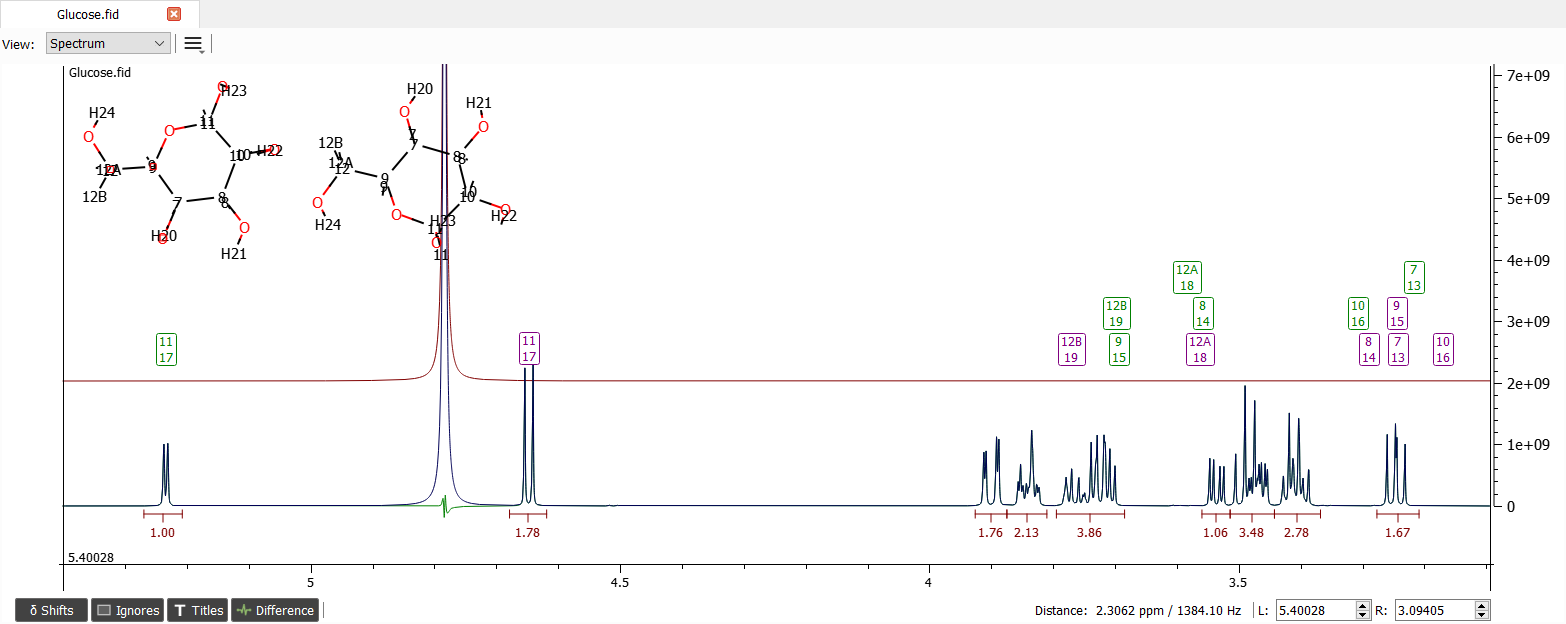
Now we may want to determine the molar ratio of the β-glucopyranose and α-glucopyranose by anomeric proton signals to ease the analyzation of the spectrum. Let's zoom into the anomeric proton range and iterate with the 'Total Line Shape Fitting':
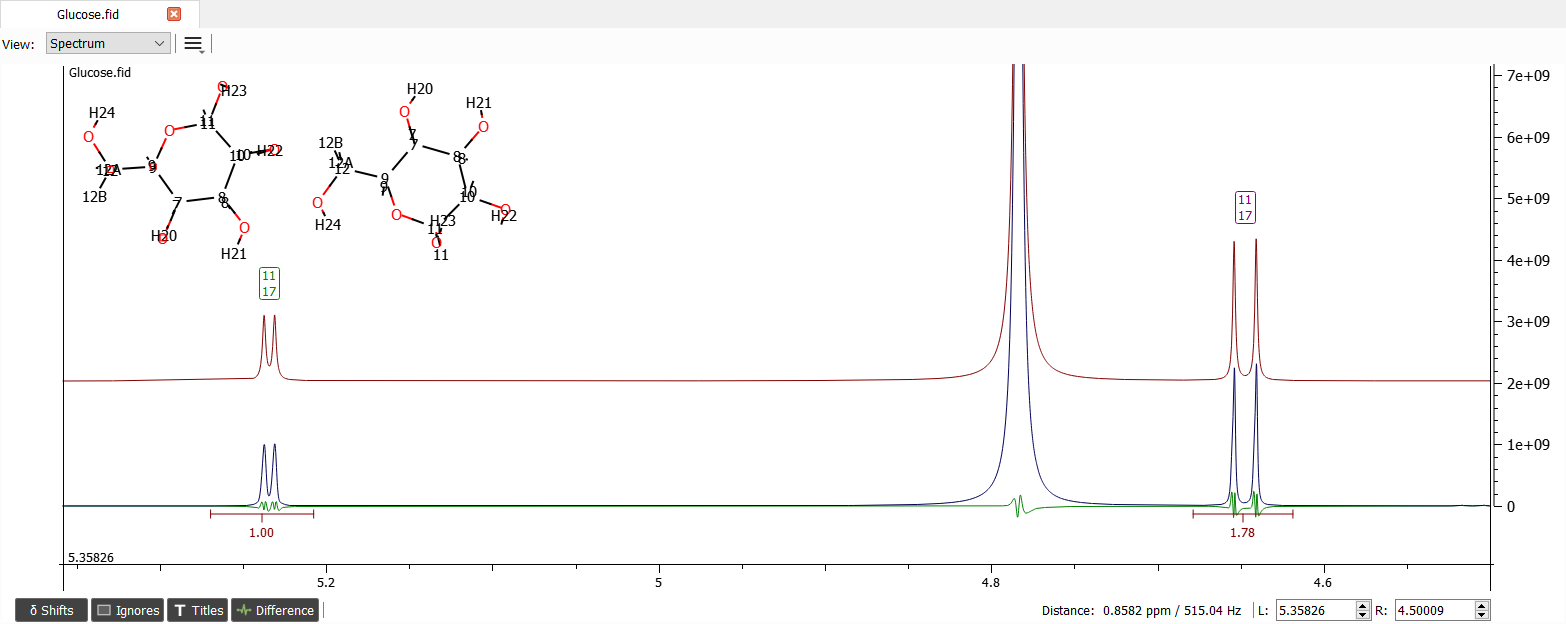
Note that the molar ratios may not be the final ones if the signals overlap with another isomers of glucose, but these are very good guesses. Now we can zoom into the region of 3 to 4 ppm:
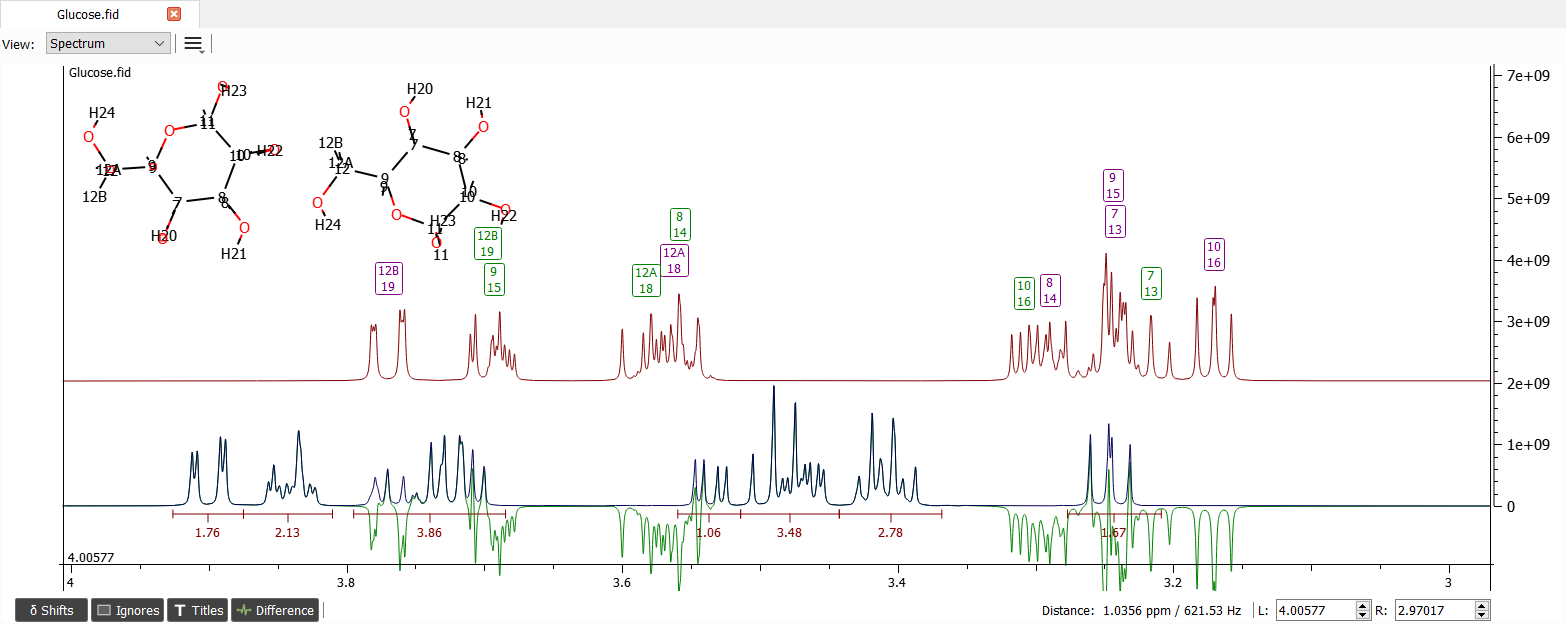
There seems to be some systematic error. Let's choose all of the shift labels in this area by dragging mouse over the labels:
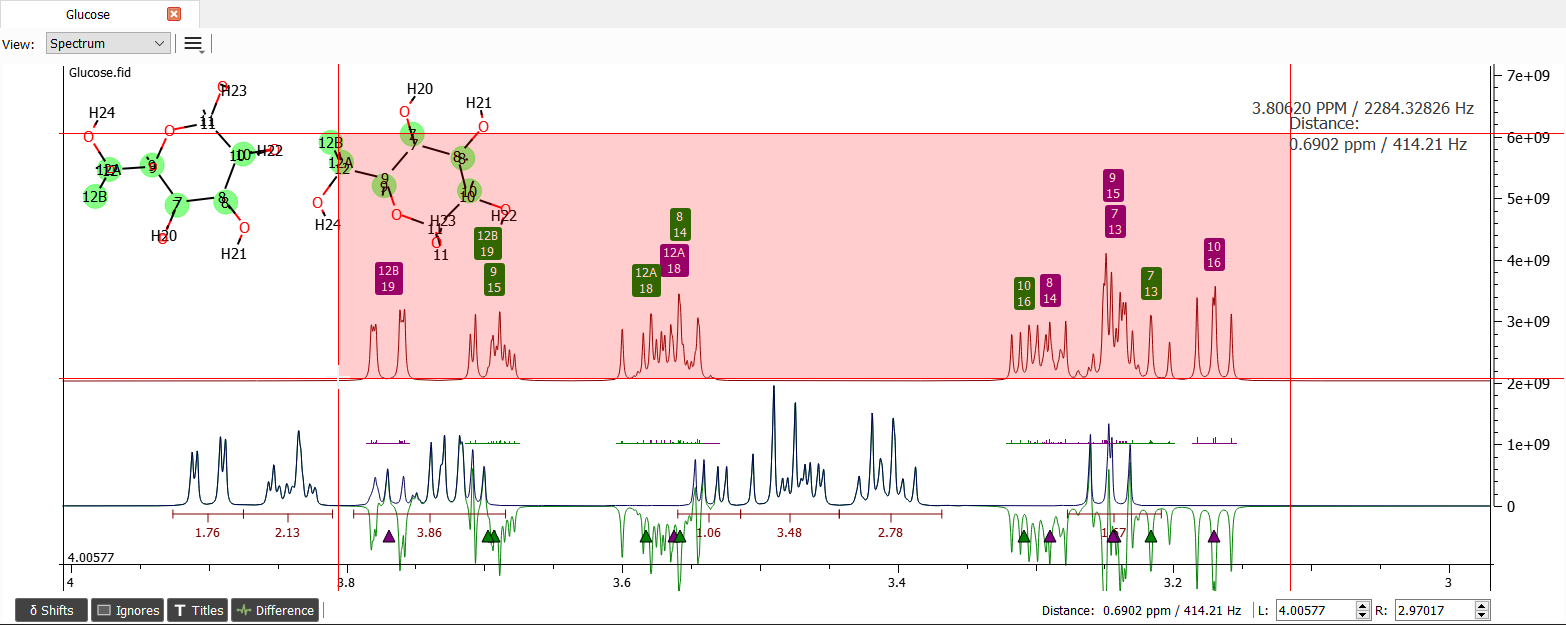
When the labels are moved and fitted by the most left one signal, the spectrum will look like this:
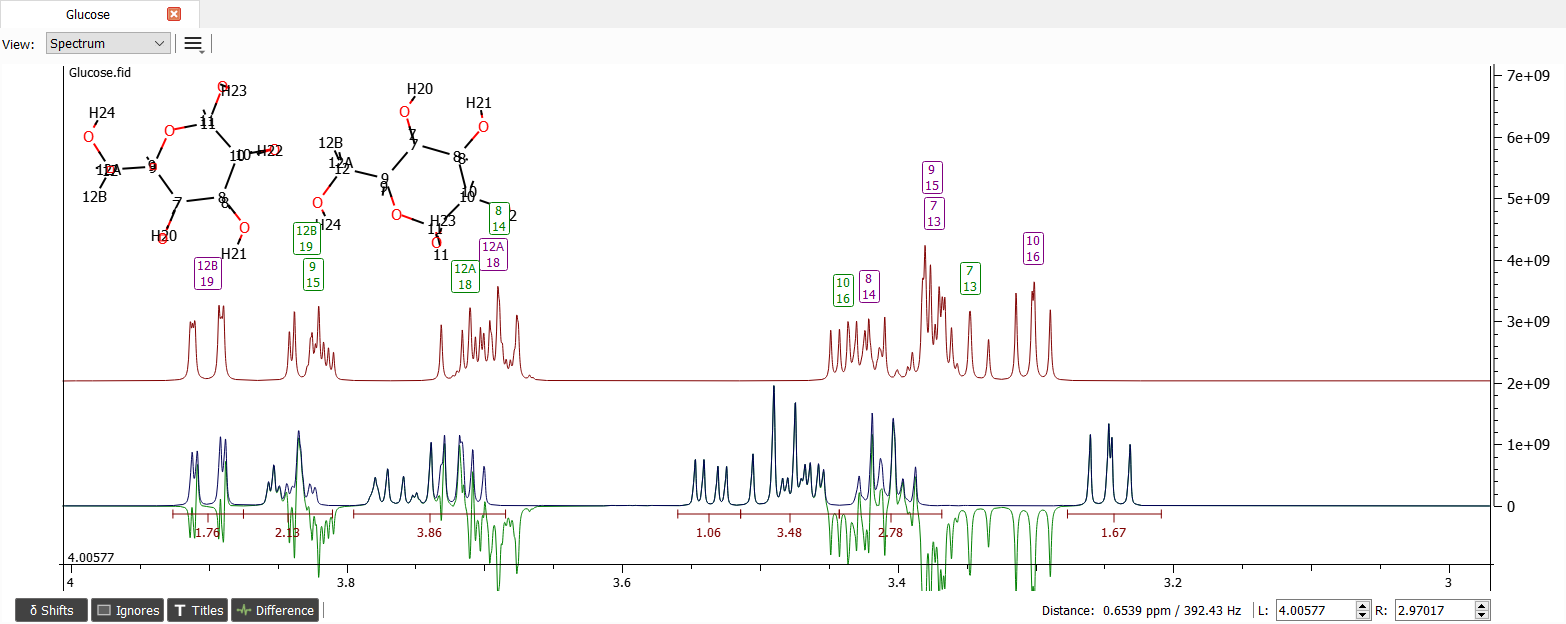
Now the signals are a bit easier to move individually to correct or almost correct positions:
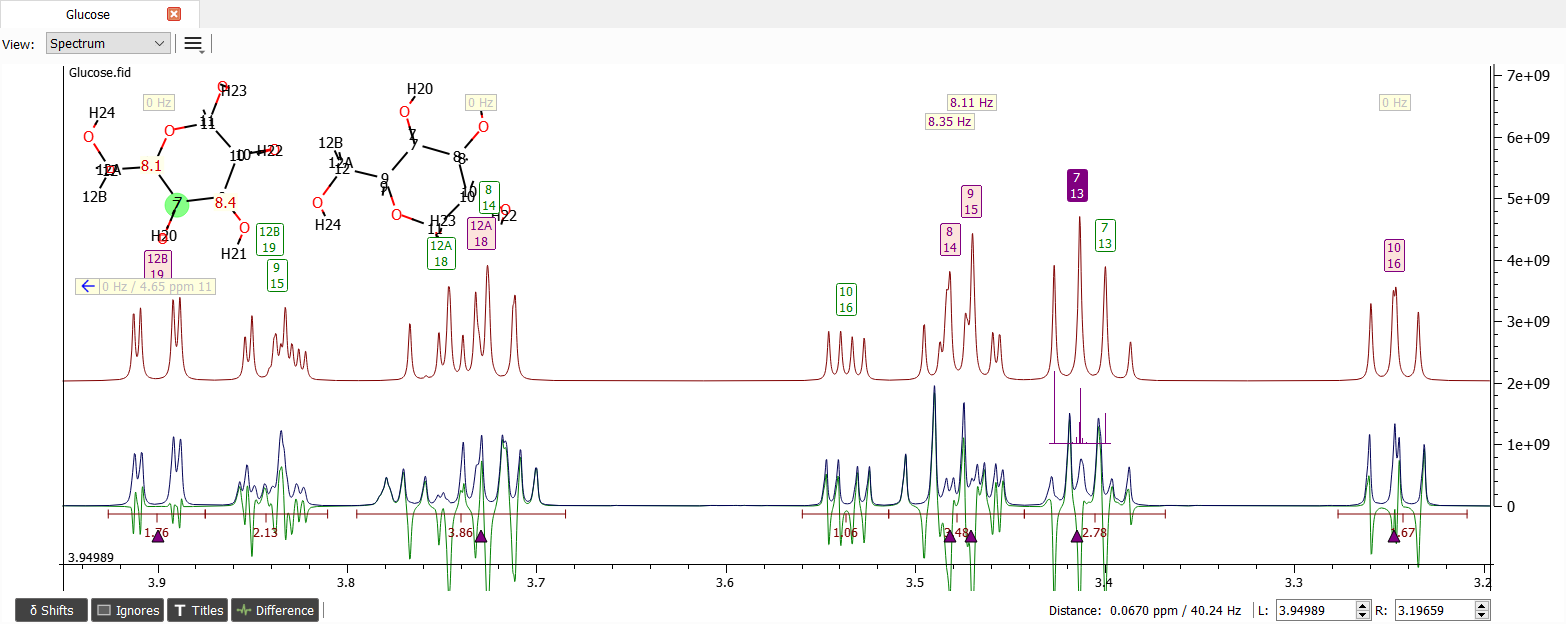
Now the shift '7' of both isomers seems to be in wrong order. Also the shift label '8' of β-glucopyranose needs to be moved a bit to the left:
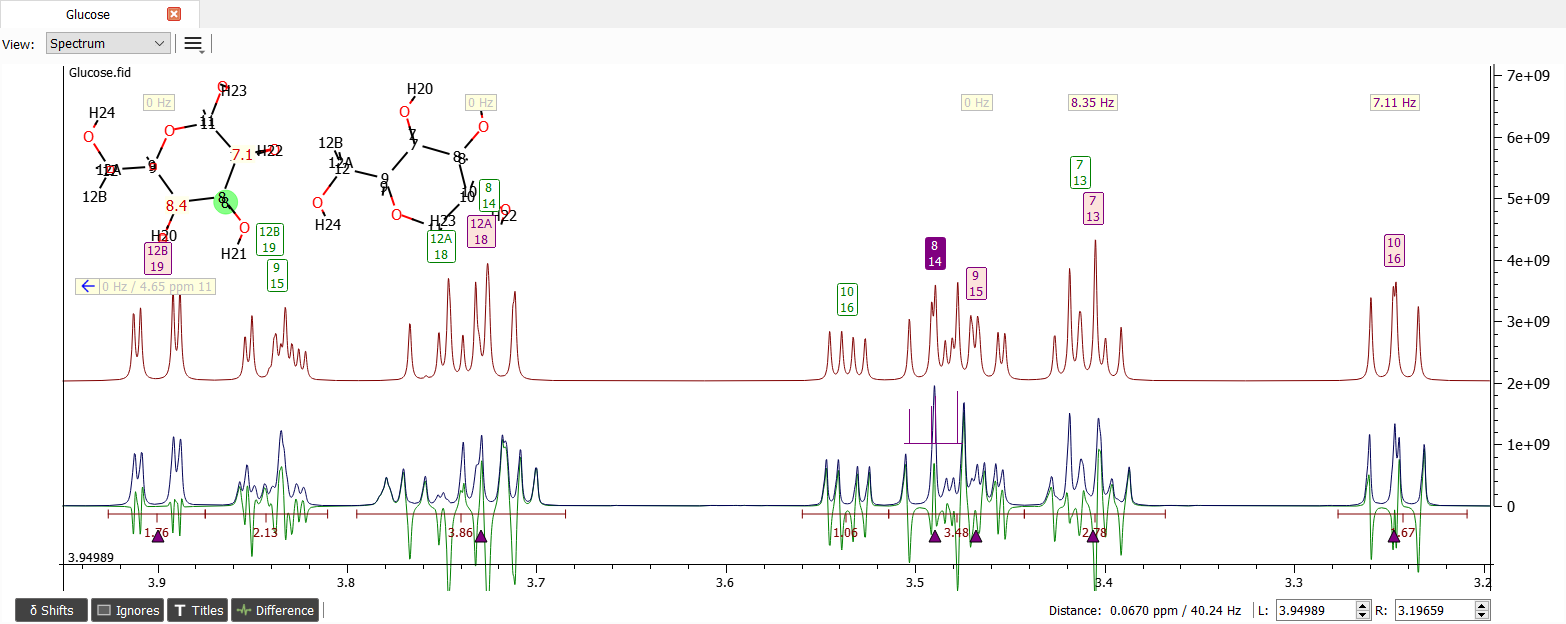
The region from 3.2 to 3.6 ppm seems to be quite okay. Let's zoom into the region and iterate:
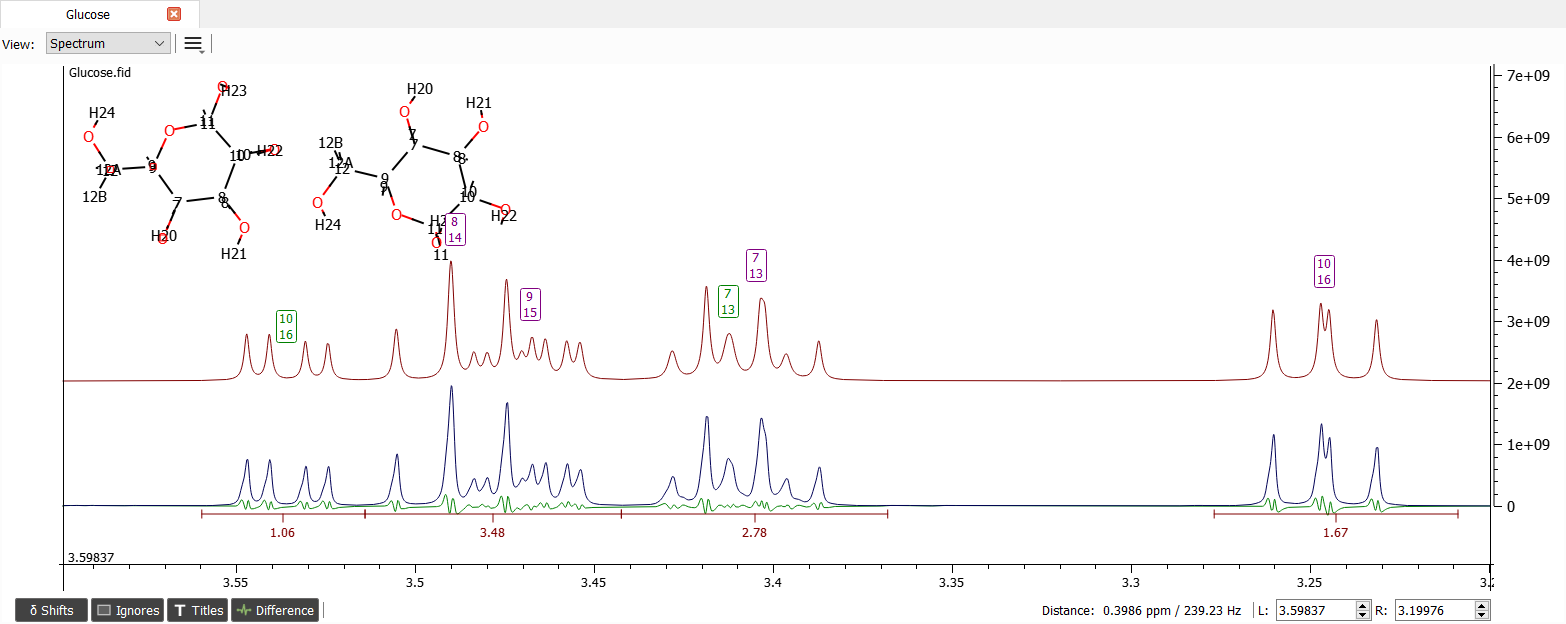
After zooming into region from 3.6 to 4 ppm and moving the shift labels to close correct positions the spectrum looks like:
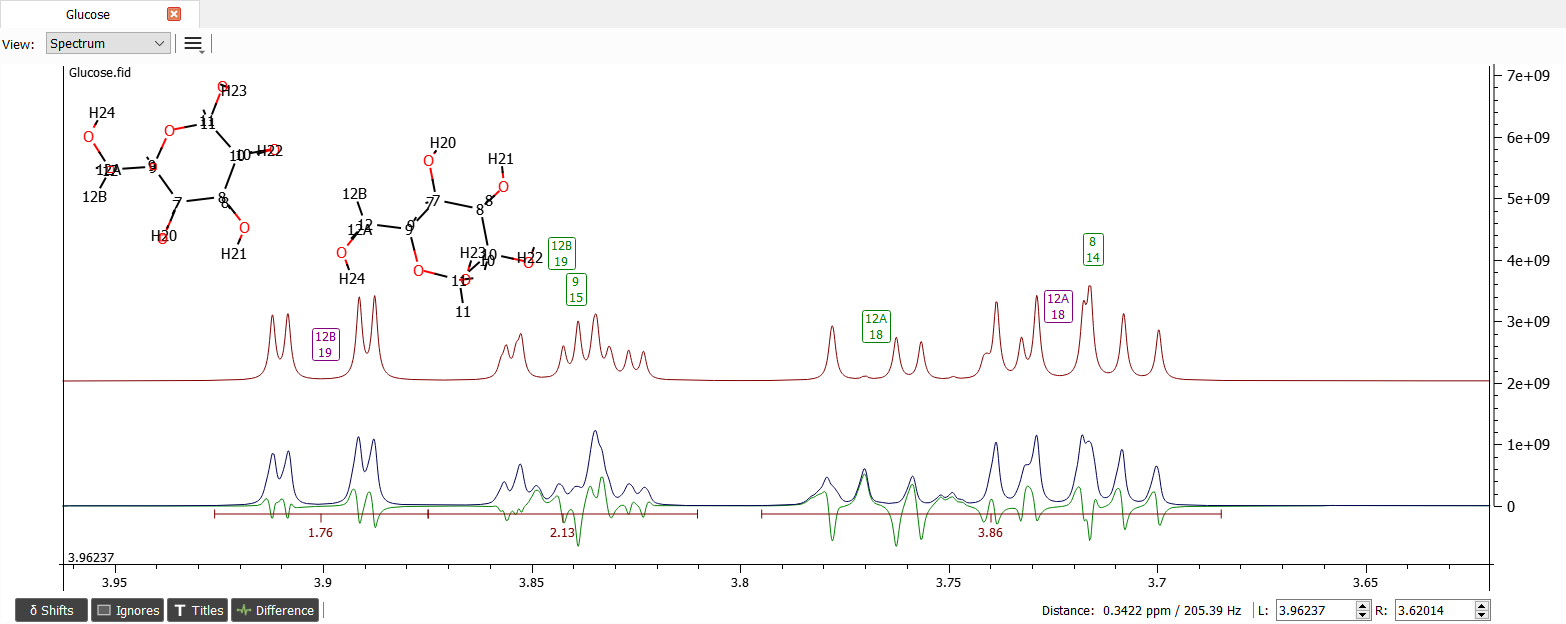
Because the other Nmr parameters are quite okay let's zoom out to region from 3 to 5.4 ppm and iterate the spectrum:
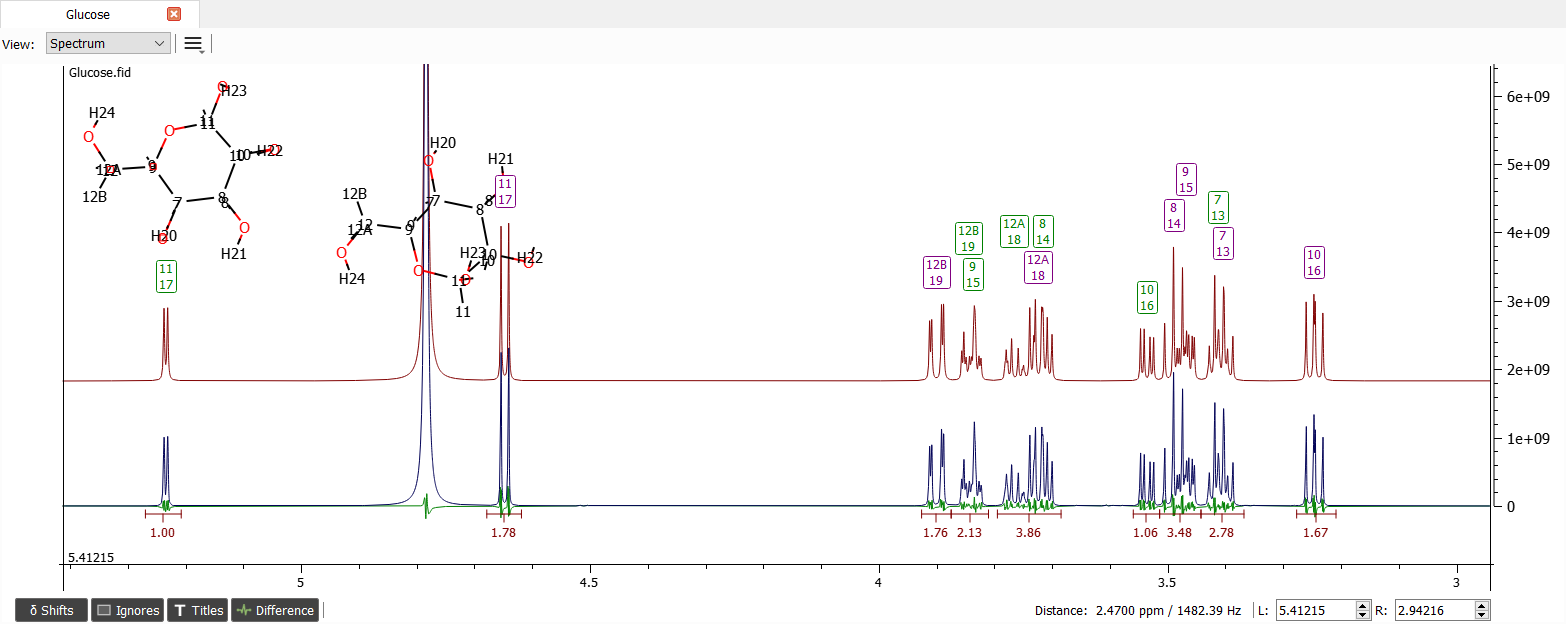
This is enough for us. Lineshape could also be iterated and long-range couplings found, but those are quite irrelevant for this tutorial. The final concentrations are:
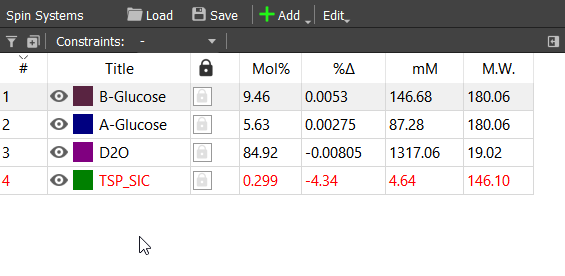
From the concentrations of α- and β-glucose we can calculate that the molar ratio for these isomers are approximately 37.3% and 62.7%, respectively. We can also seek strategy for determining molar ratios of less abundant isomers. This is very hard problem directly from this spectrum, but it is possible by using parameters of these isomers. These parameters could be obtained with 1D-TOCSY experiment. For example irradiating the small signals at 4.25 or 4.32 ppm will give the spectrum of only α- or β-glucofuranose. The signal at 8.46 ppm can also be identified to the aldehyde proton of open chain form.