Tutorials
Intermediate tutorial 2
In this tutorial we make ourselves familiar with processing raw spectrum with ChemAdder.
Contents
Phase correction
Now we have raw spectrum which needs phase correction.
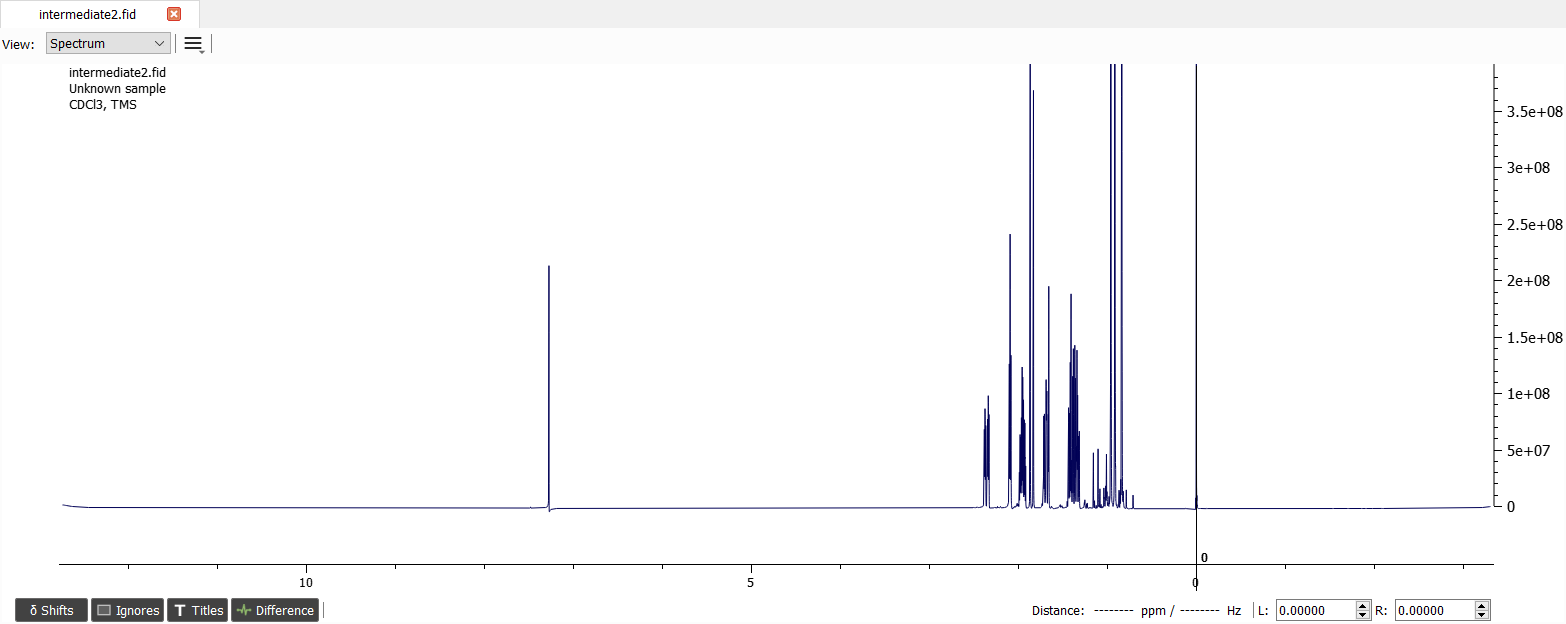
Let's start by zooming in to see the incorrect phase and choosing the 'Manual Phase Correction...' from 'Phase correction' button.
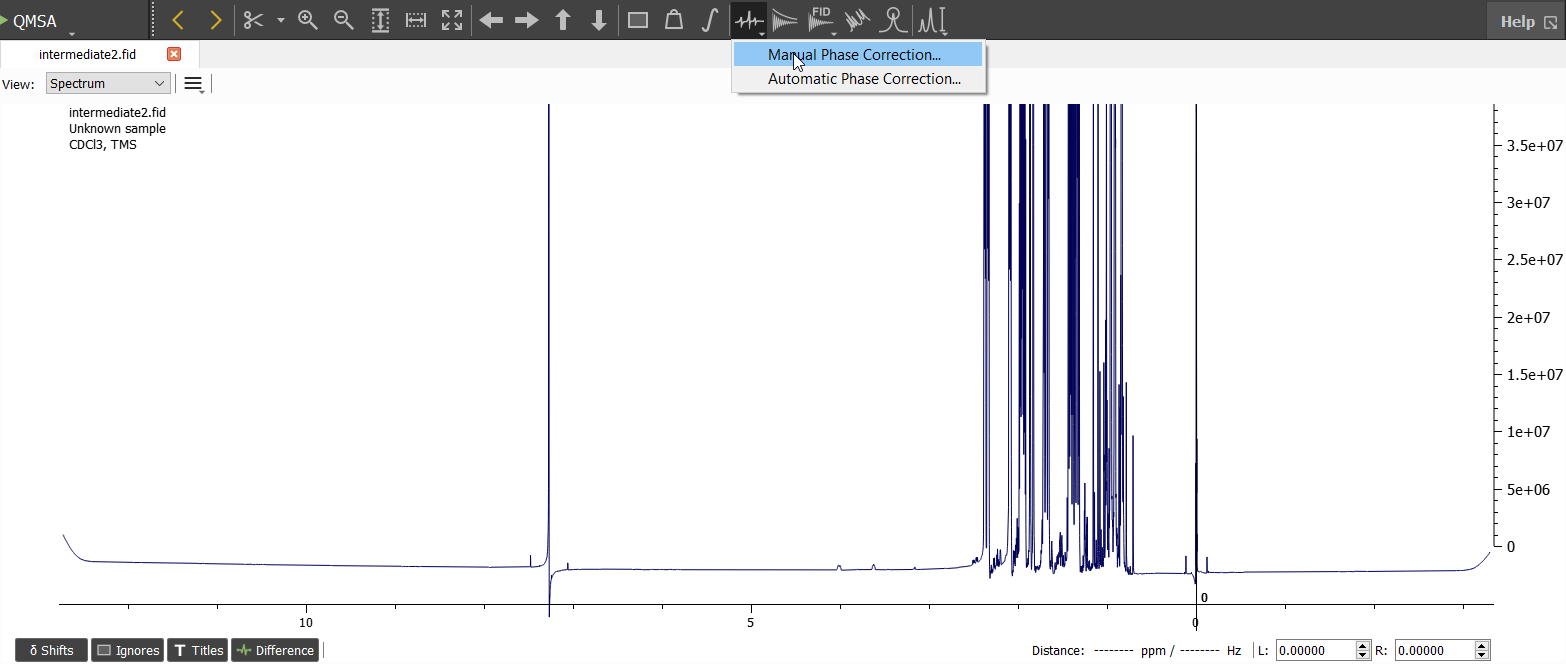
The green line is the 'Pivot point' which can be adjusted by dragging. The default position for pivot point is the most intense signal on the spectrum. This is great position almost always. The purpose of phase correction is to get the signals symmetrical and pure up signals (or down in case of e.g. Dept 135 spectrum). In this case the zeroth and first order phase correction is needed. The zeroth order phase correction can be made by dragging with left mouse button pressed down on the 'Phase Correction Window'. The first order correction can be made with right mouse button. Use also Ctrl to fine tune. The baseline seems to be negative so it's adviced to move the baseline upwards to allow zooming in and keeping the signals visible. This can be done by moving cursor on the position of zero intesity and dragging upwards (see picture below):
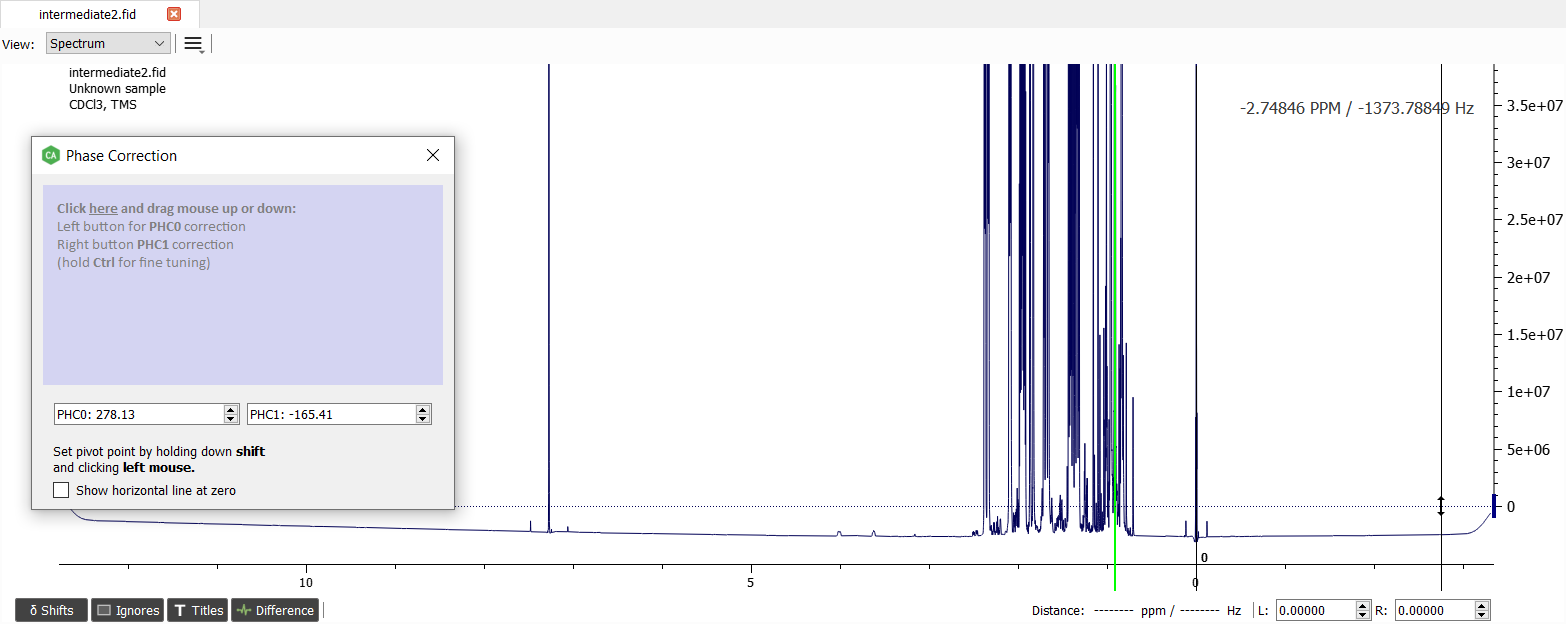
When the phase looks good enough, we can close the 'Phase Correction Window'.
Baseline correction
Next we can open the 'Baseline Correction Window':
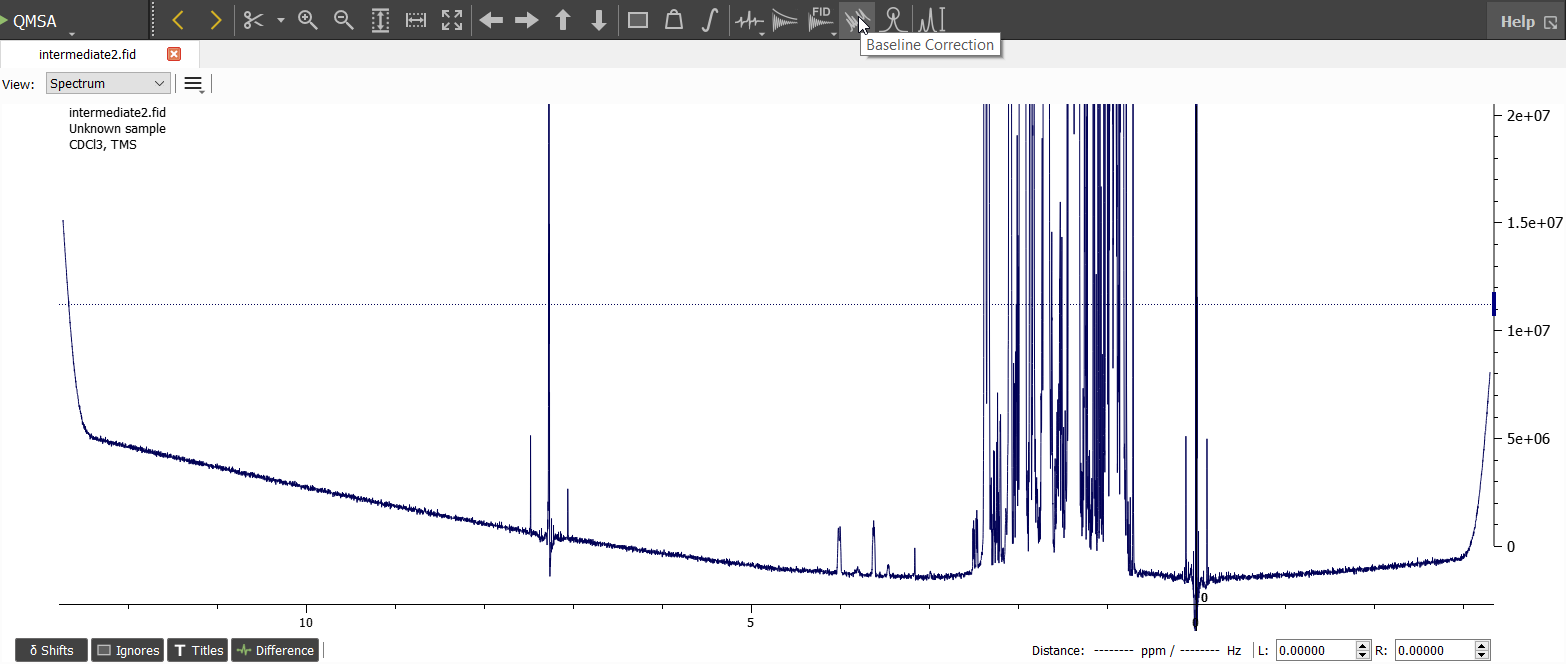
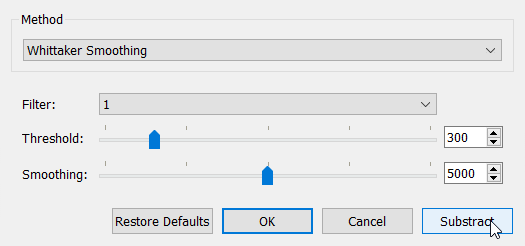
In this case the default options of 'Whittaker Smoothing' seems to be quite good. You may also change the options if you like. There is also 'Crowded Whittaker Smoothing' method which is useful quite often. When we have found good enough options, we can click 'Subtract' and accept the result by clicking 'Ok':
Let's drag the baseline back to zero intensity and rescale to full size:
Analyzation
We can ignore the solvent signal at 7.27 and reference signal at 0 ppm. Let's start the analysis by zooming into the area between 2.5 and 0.7 ppm.
Let's integrate the signals and normalize one of the methyl signals:
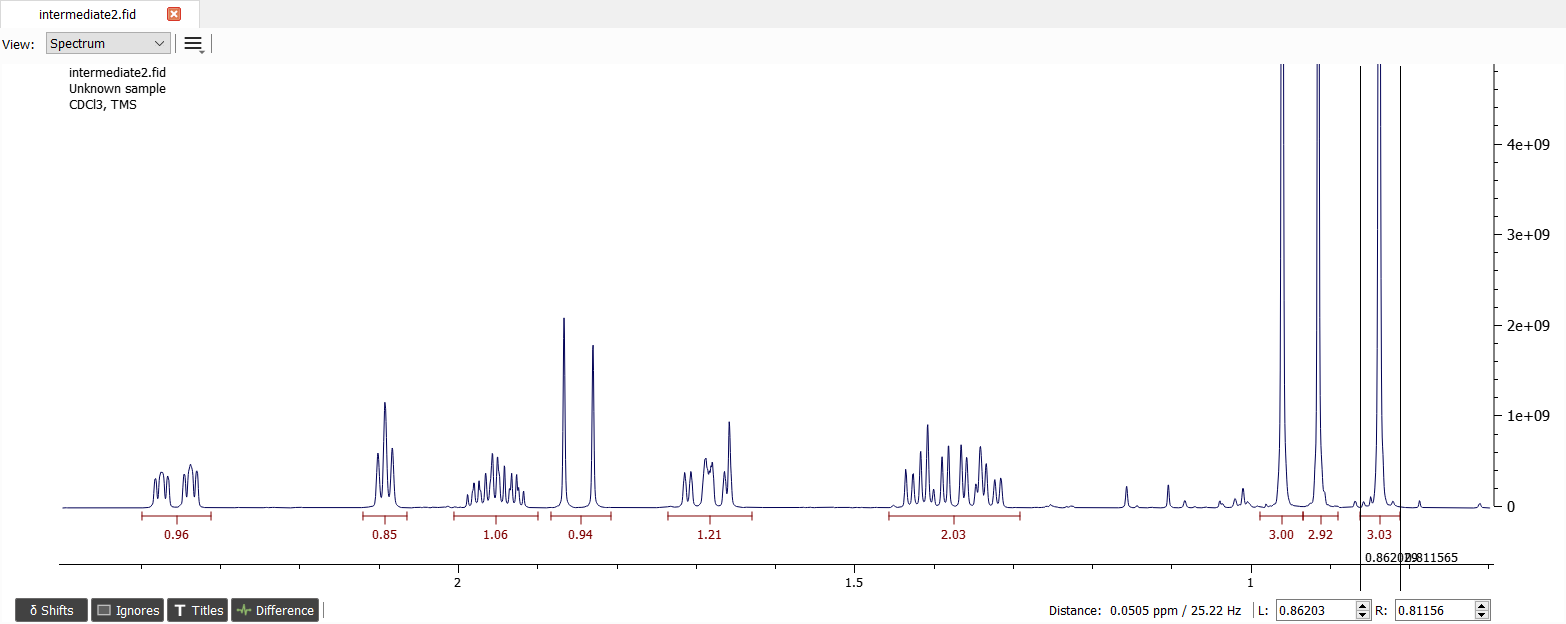
There seems to be ten signals. Let's create spin system with 10 chemical shifts and set the shifts to correct positions:
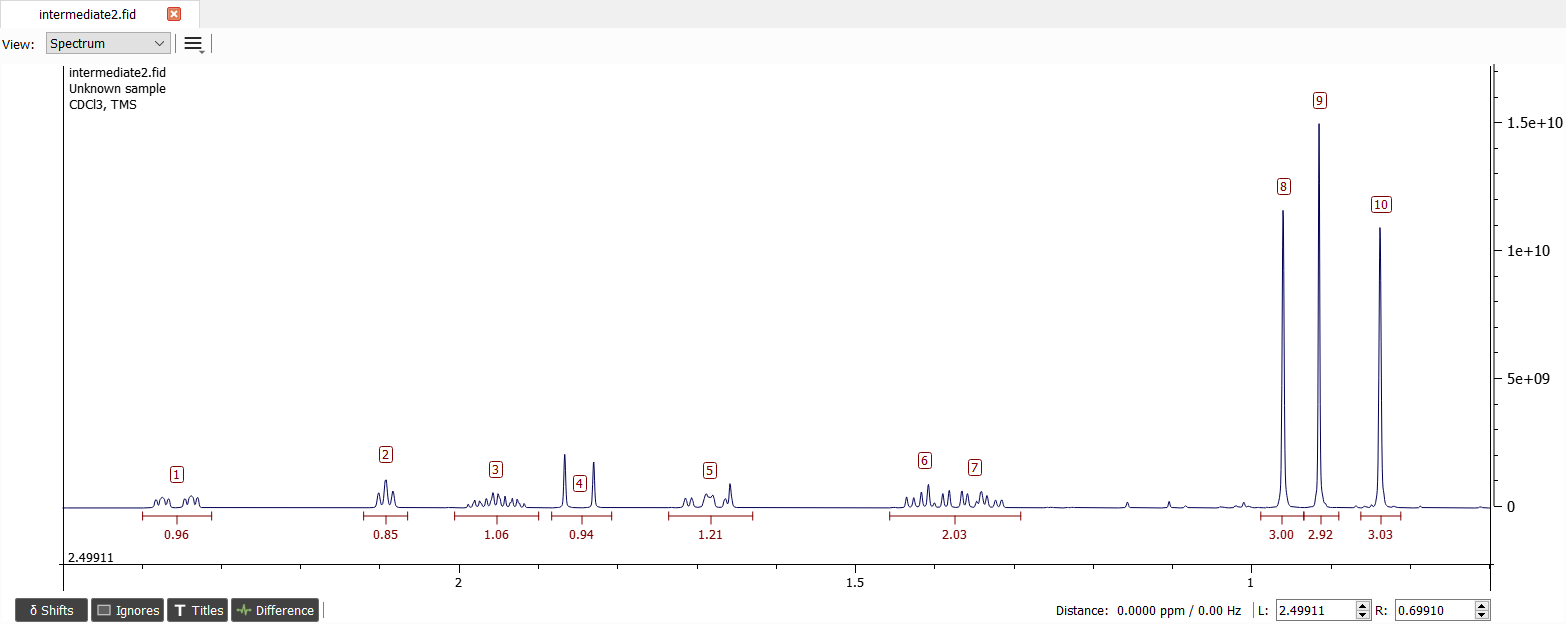
Remember to change the spin particle types for methyl signals. We can immediately see very large coupling between signals '1' and '4'. This large J-coupling is geminal coupling and there is clearly carbonyl group close by these protons. The coupling is approximately -18.2 Hz. Note that the signal '4' doesn't have any other couplings. This indicates that the angle between proton '4' and proton at three bonds away have to be about 90°. Also the molecule needs to be quite rigid. There also seems to be coupling of 4.0 Hz between shifts '5' and '7'.
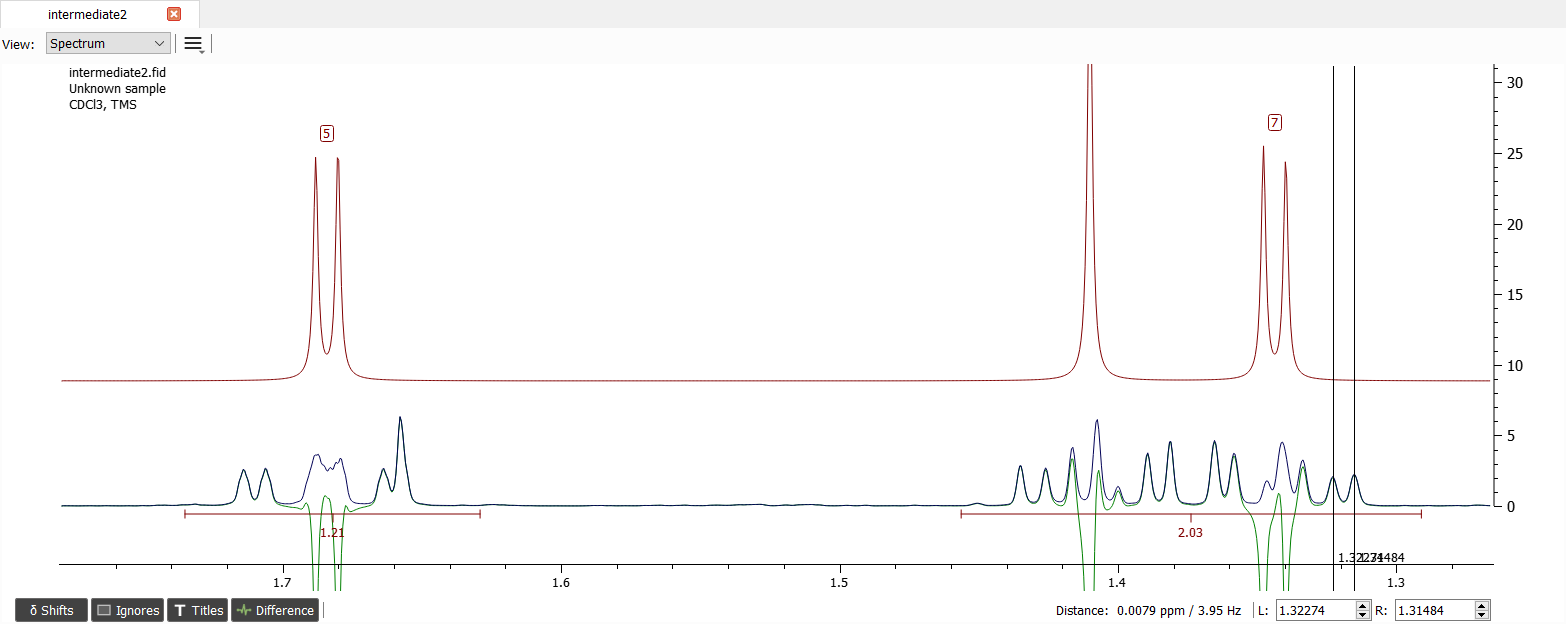
Since there seems to be some roofing effect between shifts '6' and '7', they are coupled. We can guess the J-coupling to be e.g. 8.5 Hz:
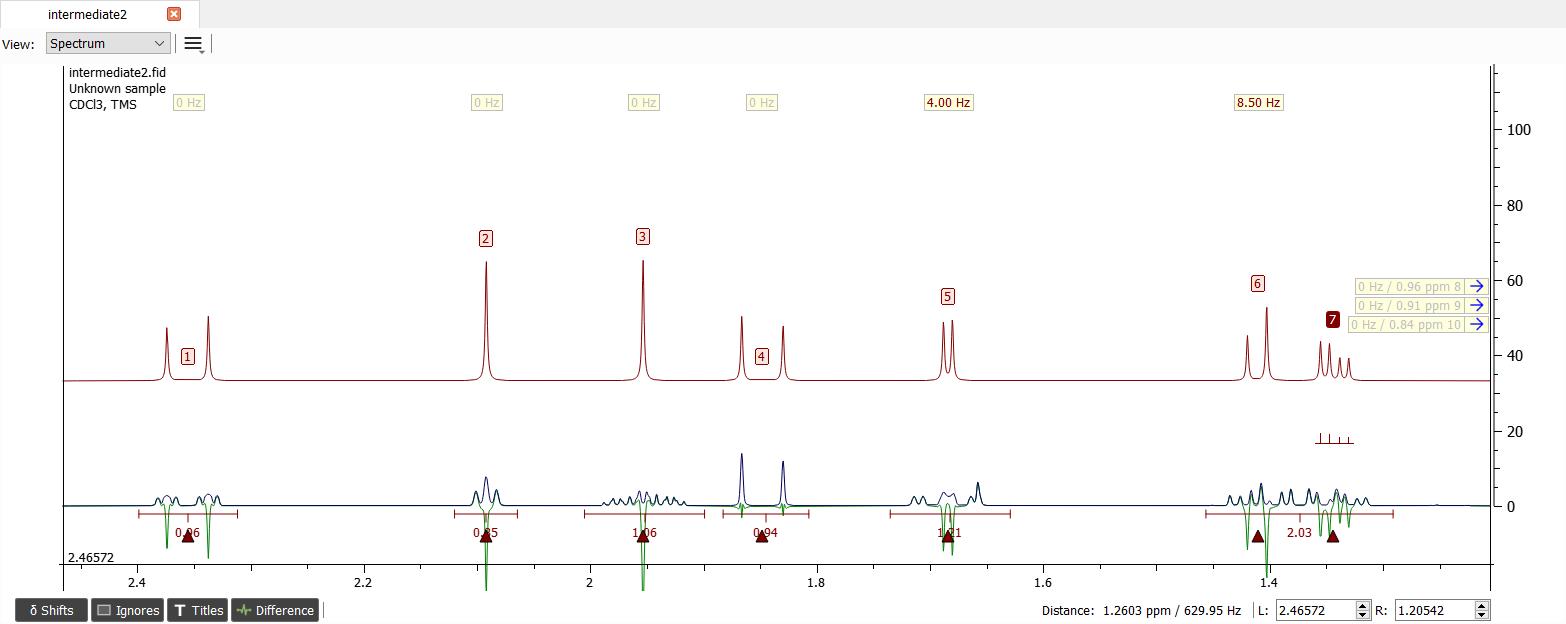
The signal '7' is still missing one large coupling. The only possible signal to be coupled with is signal '3' since others have quite small couplings and the signals '5' and '6' are already coupled with '7'. We can deduce that the signal '3' is geminally coupled hence the J-coupling is about -12.5 Hz.
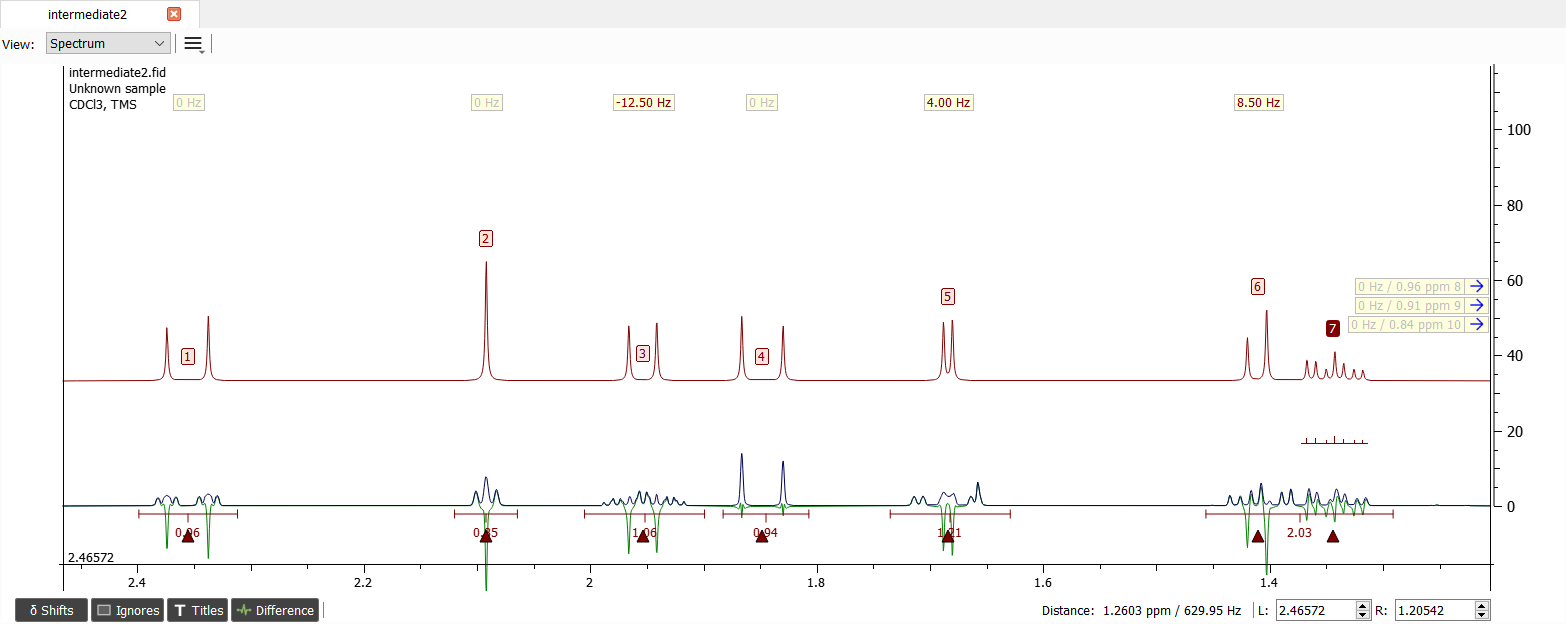
Now the signal '7' seems to be ok. The signal '6' is quite similar and needs a geminal coupling. The only possibility is signal '5' so let's add coupling of -12.5 Hz between these signals.
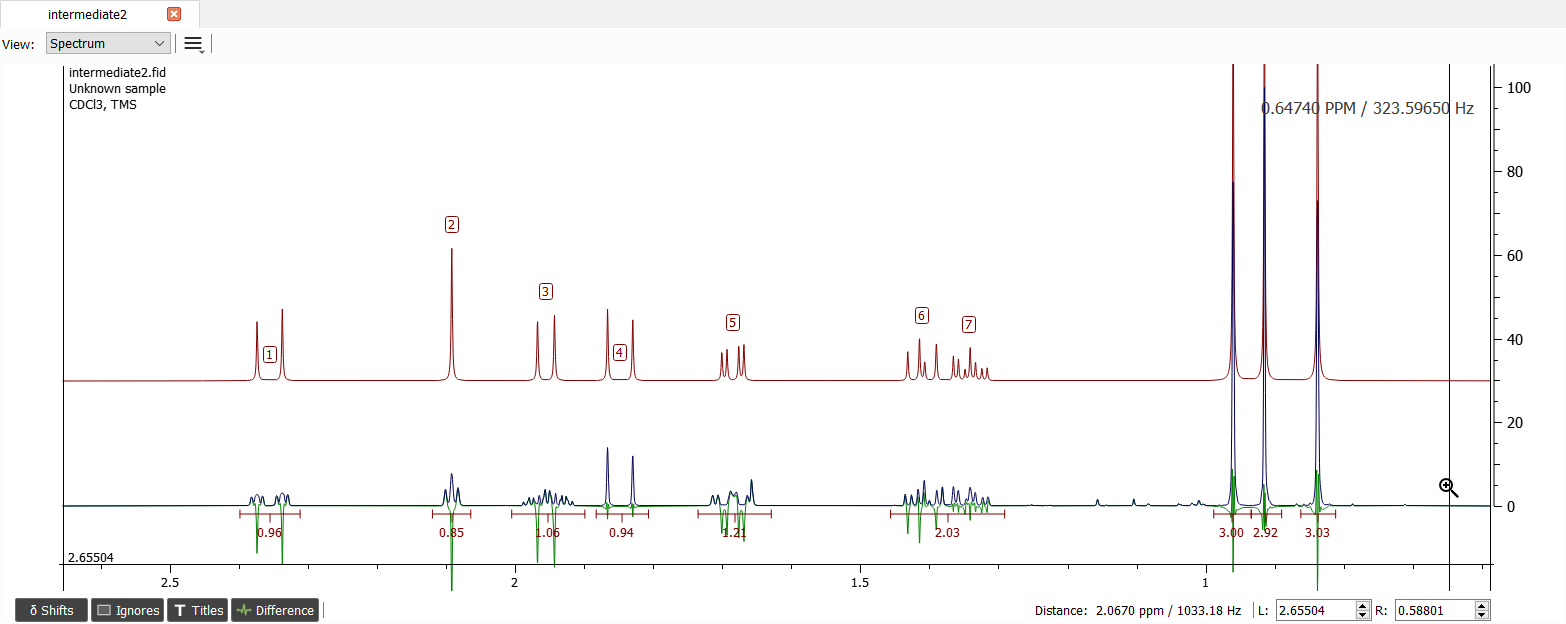
The signal '5' is also missing large coupling and it must be coupled with signal '3' since there aren't left any other signal missing large coupling. Let's add e.g. 8.5 Hz coupling between these signals.
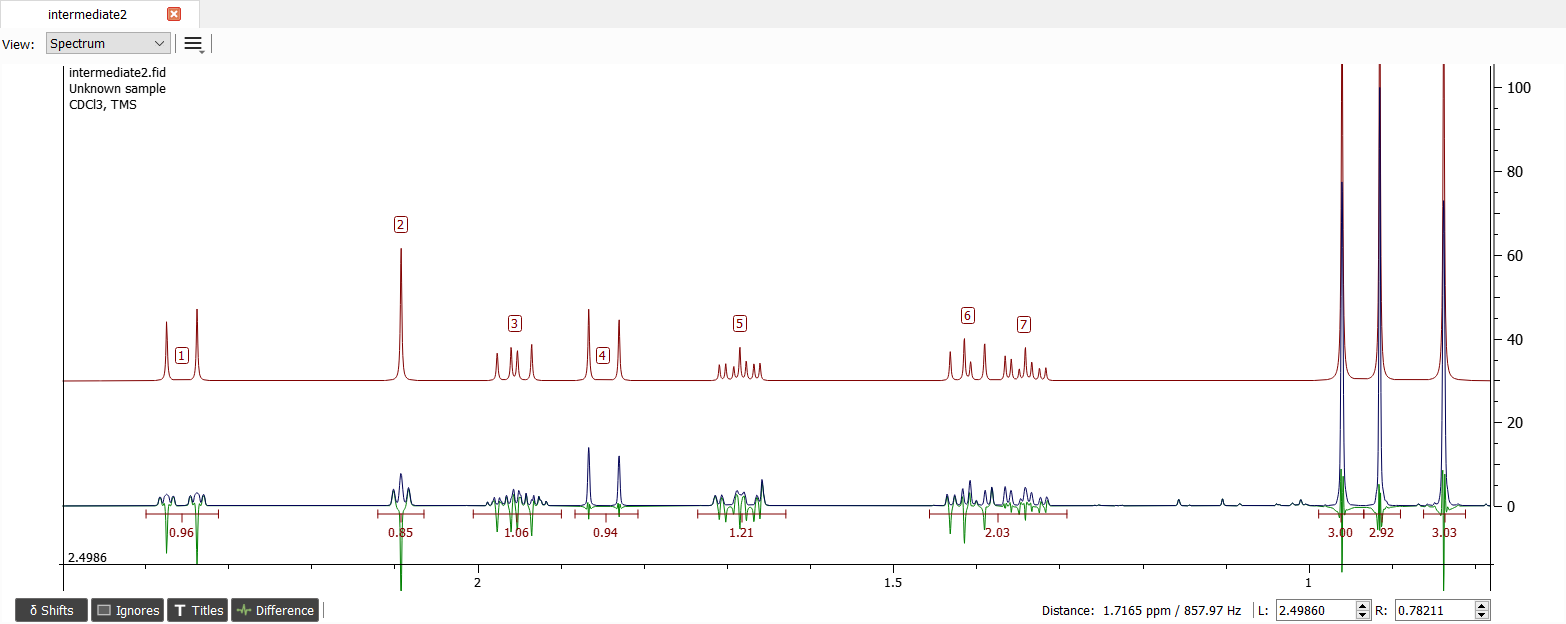
The signals '1', '2' and '3' are missing at least two vicinal couplings and the signal '6' is missing one coupling. The coupling of signal '6' is 4.6 Hz which is too large to be coupled with signal '1' hence the signal '1' is coupled with signal '2' and '3'. Those couplings seems to be about 3.5 Hz.
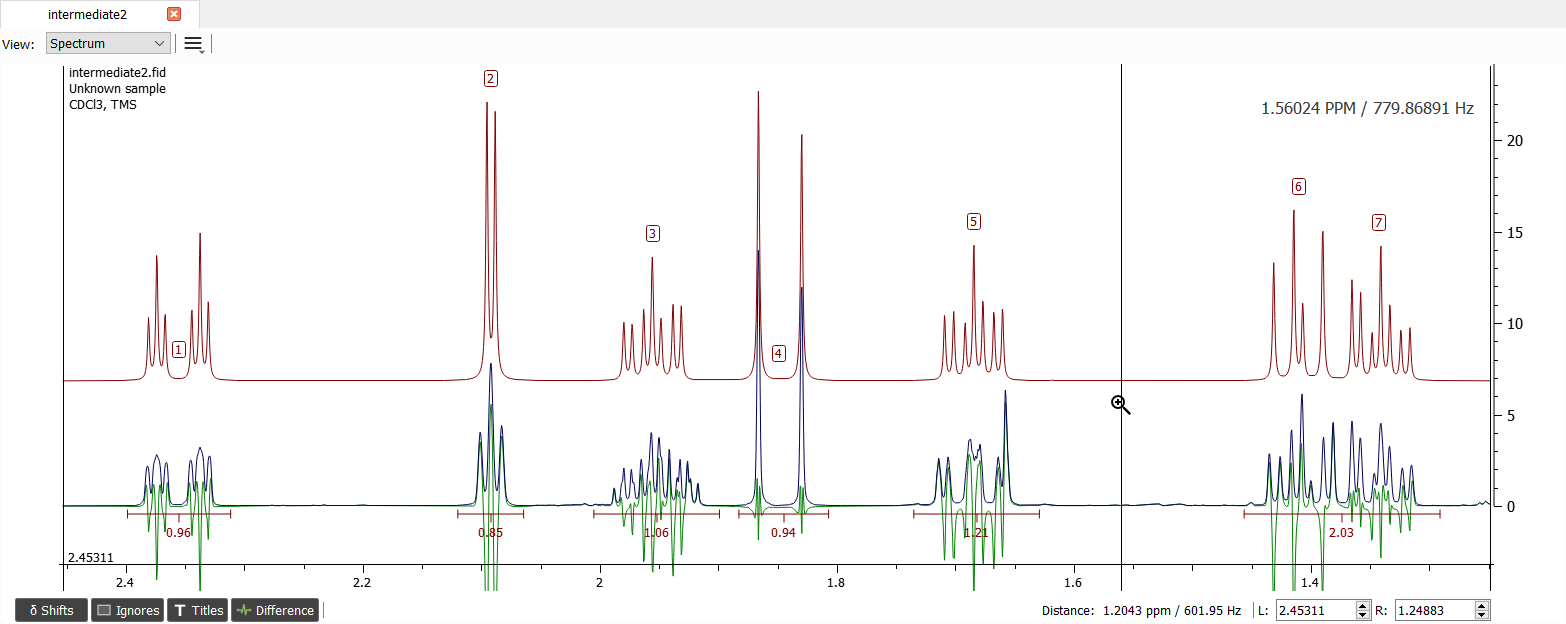
The signals '2' and '6' are missing one coupling and the signal '3' is missing one or two so the only reasonable couplings are between '2' and '3' as well as '3' and '6'.
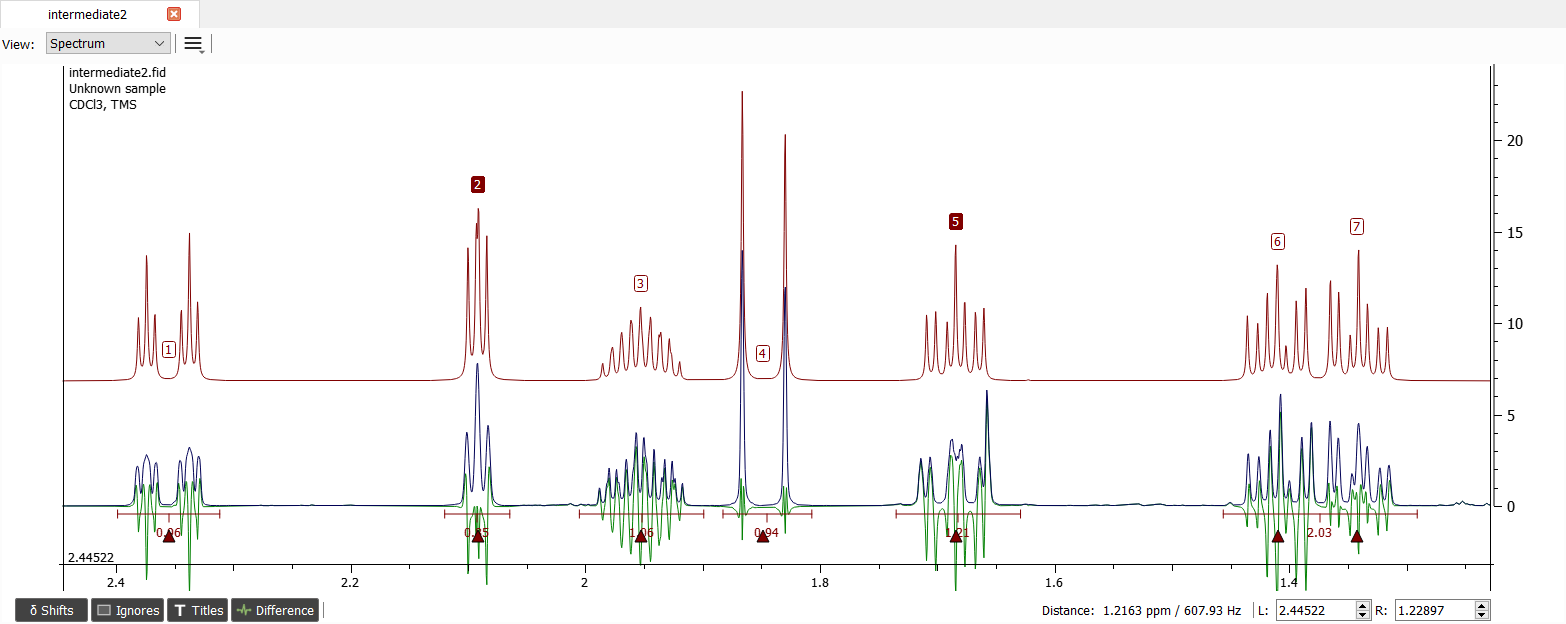
Now we can let the iterator do the job:
We could continue to enhance the lineshape or adding long-range couplings but the spectrum looks good enough for solving the molecule structure.
Structure determination
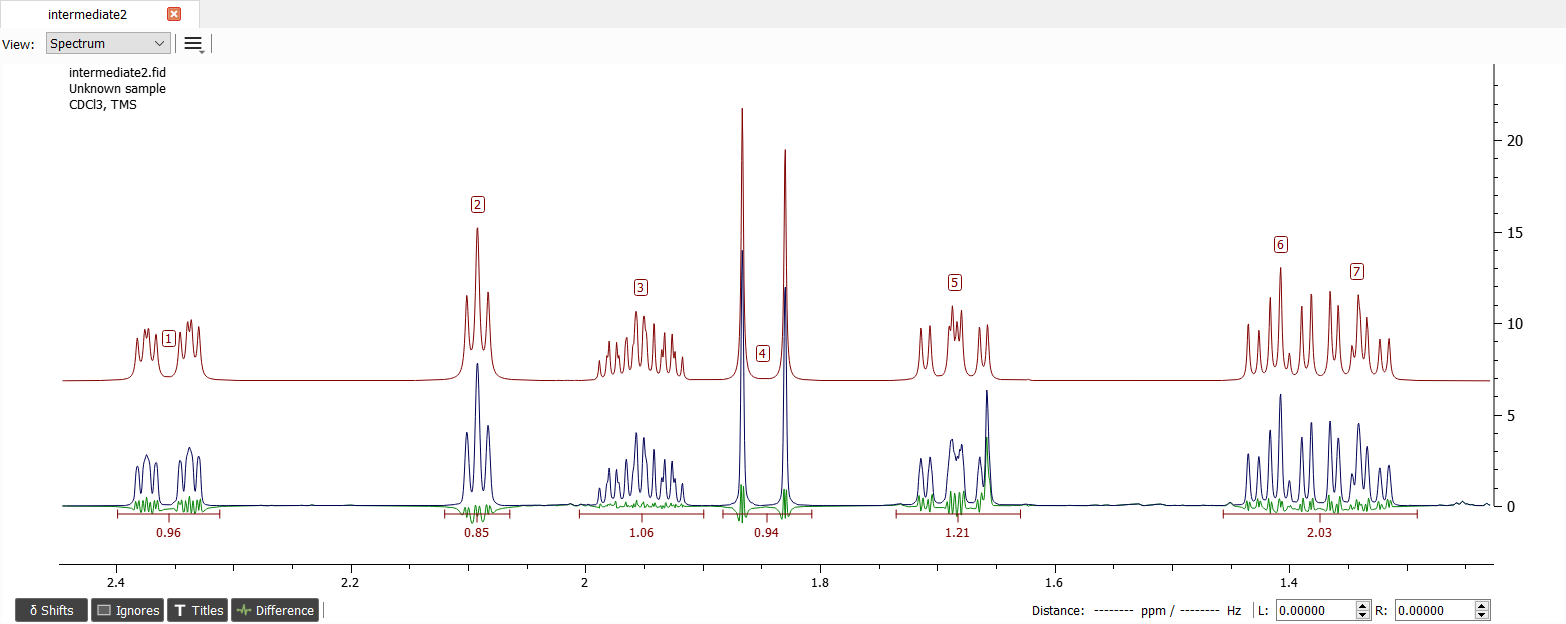
Let's start with the signal '1' and '4'. It was mentioned before that this large coupling between the signals is geminal and there is a carbonyl group close by. There was also small coupling to signal '2' and '3', but the latter one is in fact 4J-coupling.
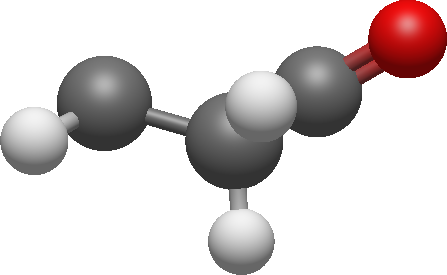
The signal '2' was coupled to signal '3'. We also found that signal '3' was geminally coupled to signal '7':
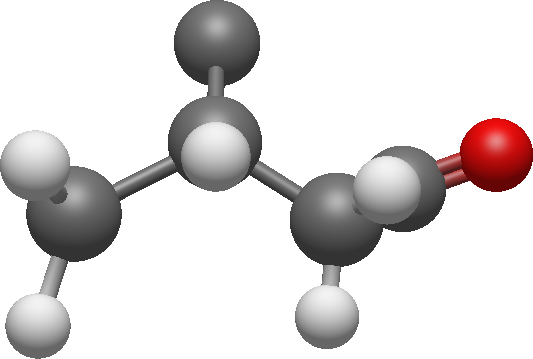
The signal '7' was also coupled to signal '5' and '6'.
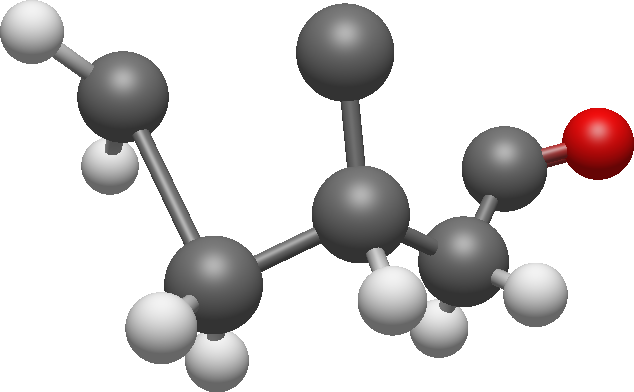
There aren't any other couplings so we can add one carbon and connect the carbons:
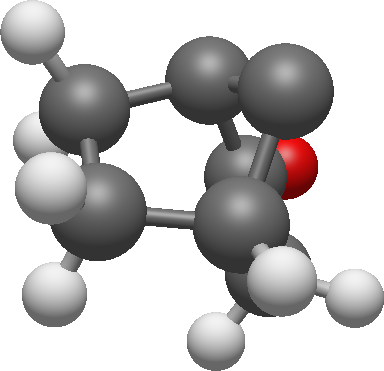
Now we can add methyl groups and assign the last signals to the structure:
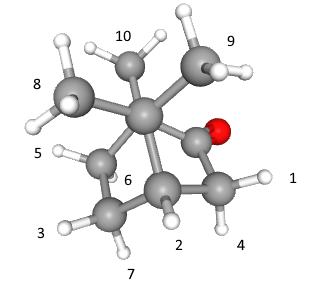
The molecule is camphor.