Tutorials
Basic tutorial 1
In this tutorial we learn basics of spectrum analysis with ChemAdder.
Contents
- Creating spin system
- Spin particle determination
- Shift and proton assignments
- Adding J-couplings
- Spectrum simulation
- Tip: Show only coupled shifts
- Parameter iteration
Creating spin system
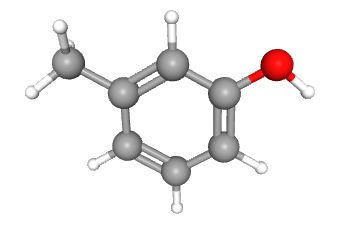
In this section we will learn how to create spin system manually. Let's start with a simple molecule m-Cresol.
As we can see the spectrum contains reference and solvent signals along with the m-Cresol signals. The reference used is TSP and solvent is D2O. Solvent has residual HDO signal at ~4.82 ppm.

Because we aren't interested in quantification in this tutorial, those signals can be ignored setting the vertical cursors around the signals by right clicking and pressing the 'Ignore Region' button.

Now we can create spin system for the m-Cresol.
Press the 'Add' button and choose 'Create New Spin Systems'.

Change the number of shifts to '5' and system title to 'm-Cresol'.

Drag the shifts (labels 1-5) one by one onto the signals.

Next we can determine the particle types for each signal.
Spin particle determination
Particles are defined with the following equation
particle = incidence * symmetry * nuclei
where incidence is the incidence of the particle, symmetry is the two-fold symmetry (1 or 2) and the nuclei is number of spins. If protons are symmetric, it's always good idea to change the particle type from 1*1*1 to correspond to the symmetry. If the (two) symmetric protons are magnetically inequivalent, you should almost always set the symmetry to 2. This allows to define 3 different J-couplings including coupling between the symmetric protons. You should note that thermal motion may average the protons to be magnetically equivalent. The normal way to use the nuclei is when the protons are magnetically equivalent and attached to the same atom. Incidence is used when the protons are attached to different atoms and the protons aren't coupled to each other.
The table below shows some examples for the particles.
| Compound | Particle | Spin System |
|---|---|---|
| one proton | 1*1*1 | |
| 1,2-dicloro benzene | 1*2*1 | AA’BB’ |
| CH2 in ethanol | 1*1*2 | A2 |
| CH3 in ethanol | 1*1*3 | A3 |
| 2*CH3 in dimethyl benzene | 1*2*3 | A3A3’ |
| TSP | 3*1*3 (or 9*1*1) | |
| Triphenylmethane | 1*1*1, 3*2*1, 3*2*1, 3*1*1 |
In the case of m-Cresol we have four 1*1*1 (aromatic) and one 1*1*3 (methyl) spin particles. The particle type can be modified by clicking the shift label which opens the settings on the left.

Shift and proton assignments
Shifts can be assigned to right protons by selecting the desired shift label and the proton(s) on the structure with left mouse button while ctrl button is pressed down. Now right clicking either the label or the proton(s) opens menu for the assignment.

Assignments can also be done after the spectrum analysis when we are certain of the coupled shifts.
The correct assignments are displayed in the picture below:

Adding J-couplings
In aliphatic organic compounds, the only coupling you need to worry about is from adjacent protons (vicinal).
In aromatic compounds, however, significant splitting does not only come from ortho protons coupled to each other, but also from meta and para protons due to conjugated bonds. Thus, coupling constants are a helpful tool for deciphering aromatic regions, and are especially vital when the chemical shifts (δ) between aromatic protons are uncertain or overlapping
Charasteric aromatic couplings:
Ortho ≈ 7-9 Hz
Meta ≈ 2-3 Hz
Para ≈ 0-1 Hz
In the case of m-Cresol the following couplings need to be defined:

2 Ortho couplings: J12, J14
3 Meta couplings: J24, J23, J43
1 Para coupling: J13
Lets start by adding the first ortho (vicinal) coupling J12:
- Select the shift 1 with mouse. After this you will see gray boxes appear over the other shift labels.
- Add coupling by double clicking the gray box over the label '2'.

The following dialog should appear. Give the calculated coupling an approximated value of '8'.

Let's add the other ortho coupling by using another method for the sake of learning. Open the J-Coupling Matrix and give the J14 coupling like in the following picture:
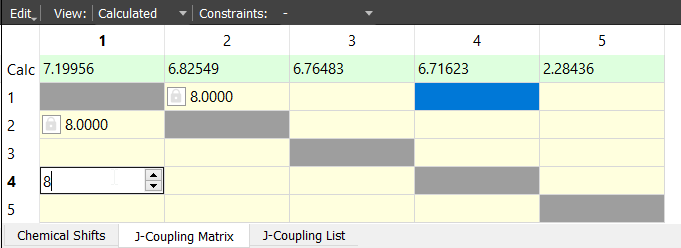
Give rest of the coupling constants by using one of the methods. The complete spin systems should look something like this:

Spectrum simulation
Now we are almost ready to simulate the spectrum. Let's add the molecular weight of m-Cresol which is 108.14 g/mol.

Simulation can be done from the QMSA dropdown button and simulate or just pressing ctrl+R.

Now the spectrum should look like this.

Tip: Show only coupled shifts
We can zoom in to the spectrum regions containing selected shift(s) and shifts which are coupled to those selected shifts by clicking 'Cut X-Axis' from the 'scissor dropdown button' (or pressing Shift+X). The picture like situation can be achieved f.e. by pressing Ctrl+A and Shift+X.
 The green line under the observed spectrum is the difference line between observed and calculated/simulated spectra. We can see that the guesses were quite accurate.
The green line under the observed spectrum is the difference line between observed and calculated/simulated spectra. We can see that the guesses were quite accurate.
Parameter iteration
Let's iterate the parameters to correct values. This can be done from the QMSA dropdown button and selecting 'Total Line Shape Fitting' or pressing ctrl+T.

After the iterator is finished the spectrum should look like this.

Normally multiple iterations may be needed before the calculated spectrum is accurate enough. Also the line shape options may be needed to modify usually but those options are discussed later. The last thing yet to be done is to assign the unassigned shifts and protons and the analysis is ready.